����һƪ����,���ڿ�ʼɸѡ�������count����
��һƪ:TCGA����GBM���ߵ�RNA-seq����
��һƪ����,���ص���ʼ����(ͼһͼ��������֮����ļ����Լ�ÿһ���ļ����е�count�����ļ�)
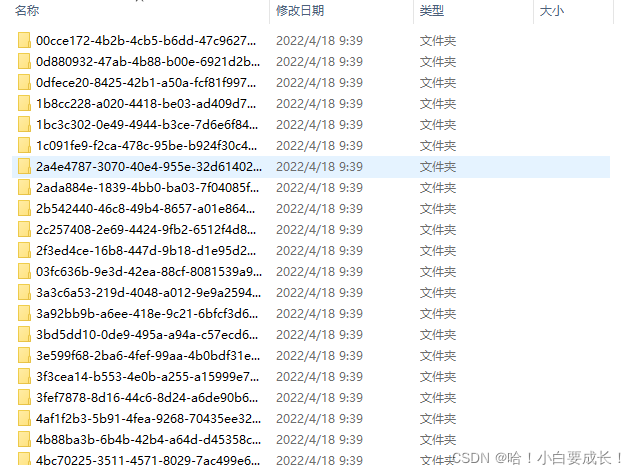

��Ҫ��ÿһ��count�����ļ���ɸѡ��gene_name��gene_typeΪlncRNA��FPKM������,Ч��ͼ����:
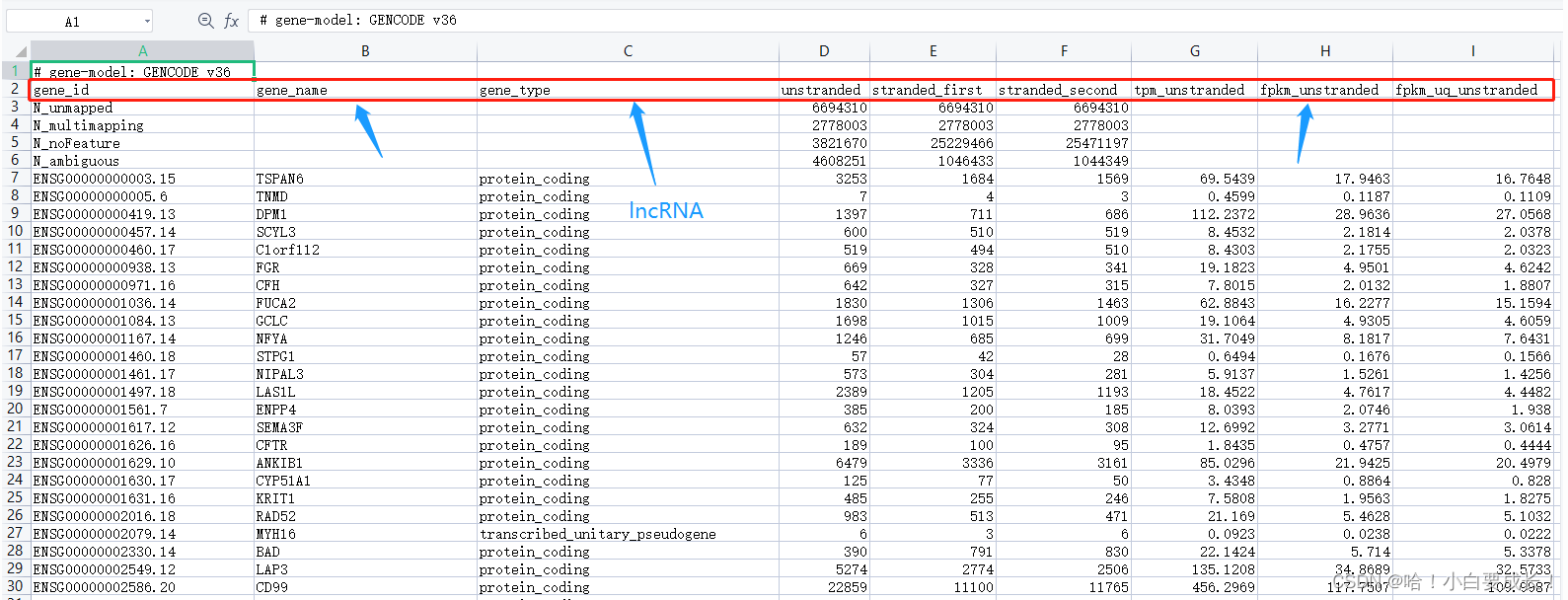


���ڲ���R����,����python��ʵ��
����:
- ��ÿһ���ļ�������ȡ����count�����ļ�,������һ�����ļ�����
- ������count�����ļ�����Ҫ������ȡ����,���ϵ�һ���ļ���
- �������ļ����ֶ�����ճ������ gene_id��gene_name��gene_type �����ݼ���(�ֶ�����,��Ϊ��Щ����һ����,����Ҫɸѡ)
1����ÿһ���ļ�������ȡ����count�����ļ�,������һ�����ļ�����
����:python | ��ָ���ļ�����ɸѡ��xml�ļ�,���Ƶ��µ�ָ��·��
2��������count�����ļ�����Ҫ������ȡ����,���ϵ�һ���ļ���
���ڲ������ tsv �ļ���������,���� tsv ת�� xls ������,����ͱȽϷ���,��Ҫ����ת����
2.1 tsv ת�� xls
������� xlrd ģ�鲻����,������´����ı�����,���Բ����� xlsx ��ʽ�����Ǻ�����ȡ�е�ʱ��,openpyxl ģ���ֲ�֧�����в���,ֻ����Ե�Ԫ��,�������ִ� xlsx ת�� xls,����һȦ,�������ӱ� (������;)��
�������ת xls �� xlsx �����õĴ���:
����:python | ������ tsv �ļ�ת�� xls �ļ�,���浽��·��
������Ҫע��,����pandas������֧��xls,Ҳ����˵����ʹ�� pandas ����Ϊ xls ��ʽ��,���� xlsx �������á�֮�������ҪѰ����������,���� tsv-xlsx-xls ���ת����
2.2 ��ȡ��ÿһ�� xls �ļ��е�������
����˵һ��,��Ϊʹ�� xlrd ģ��,��ֱ��ͨ�� sheet.row[i] �� sheet.col[i] ��ȡ�к��е�����,����ʹ�� xls ��ʽ������ openpyxl ģ��ֻ�ܶԵ�Ԫ�����,������,���Բ��� xlsx ��ʽ��
����:python | ������ȡ��ÿһ�� xls �ļ��е�������,������������,���浽ͬһ���µ� xls �ļ���
��һƪ��д������,��һƪ����ǰ�Ĺ���,ʵ��Person��ط���,����ʵ�ֹ��������繹��

