下载相关软件包:
一个python2.5版本(用conda下载不了,最低是2.7版)
配置./python2.5/Tools/Scripts到环境变量path里
windows下配置临时的环境变量,这里必须是大写的PATH:
#配置临时的环境变量:
set PATH=D:\Program Files\autodock4\python2.5;%PATH%
set PATH=D:\Program Files\autodock4\python2.5\Tools\Scripts;%PATH%
#查看环境变量变量值:
set path
#查看搜索到的python版本:
>python -V
Python 2.5.3输入set关键字可以查看当前的所有变量?
下载一个autodocksuite软件包和一个mgltools软件包,windows安装根据提示就可以。
autoduck4自带python2,估计不用自己下载python2
分子对接的步骤:
1.配置autodock的工作目录:
在cmd里配置临时环境变量PATH,打开AutoDockTools可执行文件
点击File-->Preferences-->Set?打开一个窗口:
在General菜单里,设置Startup Directory为你的工作目录
工作目录至少要有如下5个文件:
 ??将工作目录复制到输入框内,点击Make Default即可
??将工作目录复制到输入框内,点击Make Default即可

2.导入作为受体分子pdb文件、去游离O原子(水)、加H原子,保存为pdbqt文件:
选择File-->Read Molecule?将工作目录下的pdb文件导入
若是在结构预览窗里有红色的小点(游离的水(O原子)),将其删除:
选择Edit-->Delete Water
对残基加上H原子:
选择Edit-->Hydrogens-->Add-->OK
将处理好的构象选择为受体,保存为作为受体的pdbQT文件:
Grid-->Macromolecule-->Choose.. 在弹出的选择窗里选择目标对象,对出现的警告信息继续确定,对弹出的选择pdbqt文件保存路径的窗口进行确认
该步骤会自动对受体进行电荷计算
3.处理小分子配体pdb文件,保存为pdbqt文件
删掉栏目里的受体对象,同样导入小分子的pdb文件
进行加H原子操作:Edit-->Hydrogens-->Add-->OK
保存为配体的pdbqt文件:ligand-->input-->choose..?在弹窗里选择配体文件,确定后对警告弹窗进行确定。
在警告弹窗里会出现提示信息:这一步完成自动添加gasteiger电荷(修改原子类型)、合并非极性的H原子和检查结构的可扭转键。(在预览窗里结构的H数量减少)
检查配体分子的所有扭转键,调整可旋转键的位置和个数,方便之后查找配体的不同构象(半柔性对接):
Ligand-->Torsion Tree--> Detect Root (预览窗里结构出现一个绿色的小球表示这个结构的中心root)
Ligand-->Torsion Tree--> Choose Torsion (结构预览窗里不可扭转的键为红色,可扭转的键为绿色,表示酰胺键的粉色)
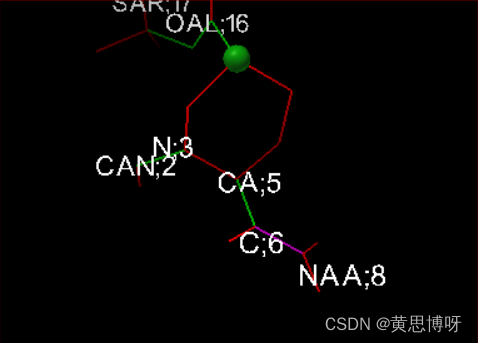
按住shift键,对粉色键进行点击,可将其转变为绿色的可旋转键,对绿色键可以做这个操作,红色键不行。
修改满意后,弹窗内点击Done确定
对配体分子保存为pdbqt文件:
Ligand-->Output-->Save?as PDBQT进行pdbqt文件保存
4.导入受体和配体的pdbqt文件:
选择Grid-->Macromolecule-->Open..?打开准备好的受体的pdbqqt文件,对弹出选择Yes
选择Grid-->Set Map Types-->Open Ligand..?打开准备的配体的pdbqt文件
方便识别,修改受体和配体的显示方式:

?受体以二级结构的形式展现,配体以键线式展现:

5.设置工作盒子
选择Grid-->Grid Box..?在预览窗里出现一个三色盒子,在弹窗来修改盒子的3轴,让盒子包围受体和配体,也可以只是包围配体和活性位点
将小分子拖到盒子外
选择DejaVu GUI?打开新弹窗:

在上面的红框内,取消mouse?transforms?apply?to "root" object only?选择
点击下方红框内的ligand将指定鼠标的操作对象
在上方红框内,?我们知道鼠标左键进行旋转,右键进行移动,滑轮进行缩放。
右键预览窗里配体移出盒子
原先取消mouse?transforms?apply?to "root" object only?重新勾选,关闭弹窗
6.保存调整好的盒子为gpf文件:
在Grid窗口内选择File-->Close saving?current
选择Gird-->Output-->Save GPF..进行文件命名和保存
选择run--Run AutoGrid?打开新的弹窗

?保证Working Directoory(wd)是我们的工作目录(由于工作目录里夹了Program File,结果计算弹窗闪退,...)
在Parameter Filename选择wd里的保存的gpf文件,会在下面的Log Filename建立同名glg文件
最后点击Launch,出现新的计算弹窗,耐心等待弹窗自动消失。
在wd下蹦出很多*.map文件
7.进行分子对接计算,结果查看和保存:
选择大分子受体:

?由于我们进行半柔性对接,所以作为受体的大分子是刚性的(Rigid)
选择配体小分子:

?选择作为对接配体的pdbqt文件,弹窗中选择accept
选择合适算法:
Docking-->Search Parameters -->Genetic Algorithm -->打开的弹窗中选择Accept
Docking-->Docking Parameters-->打开的弹窗里选择Accept
Docking-->Output-->Lamarckian GA 4.2,,-->定义dpf文件名,作为对接结果的保存文件
运行Dock计算:
Run-->RunAutoDock-->弹窗里确保工作目录正确,brower定义的dpf文件,选择launch
同样等待弹窗自动消失。
查看计算完成得到的对接构象:
Analyze-->Dockings-->Open...,打开计算完成的dlg文件,读取获取的前10个对接构象
Analyze-->Macromolecule-->Open..自动打开对应的受体大分子
Analyze-->Conformations-->Played,ranked ?by energy...,打开一个播放10个构象的弹窗
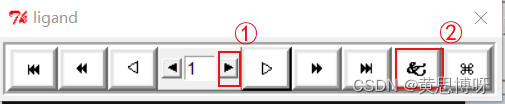
①点击播放10个构象,构象的顺序是按结合能从小到大排列
②点击打开查看H键等信息的弹窗?

?①查看该构象结合能的大小和各个能量的贡献和形成H键的个数和对应的残基原子
②勾选后在结构显示窗里可视化H键
对对接结果进行保存:
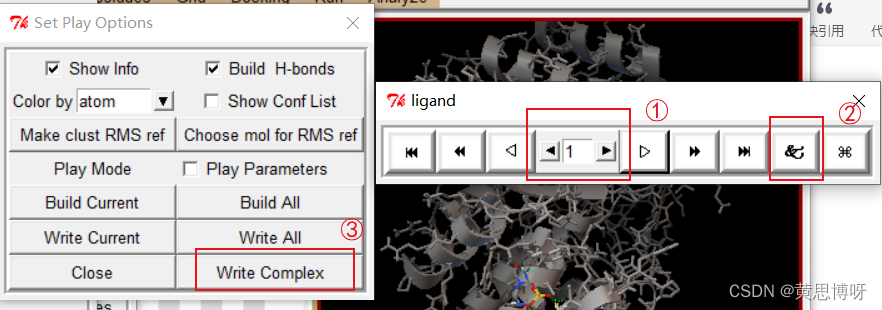
①选择第一个对接结果,该将结果的结合能最小
②点击该按钮,出现如图的Set Play Options弹窗
③点击Write Complex按钮,对第1个对接构象保存为pdbqt文件
参考视频: