ЮФеТФПТМ
ЧАбд
БОЮФМЧТМСЫQuantifyPoly(A)1АќЪЙгУЙ§ГЬжаЕФвЛаЉЮЪЬт
вЛЁЂЮФеТФкШн
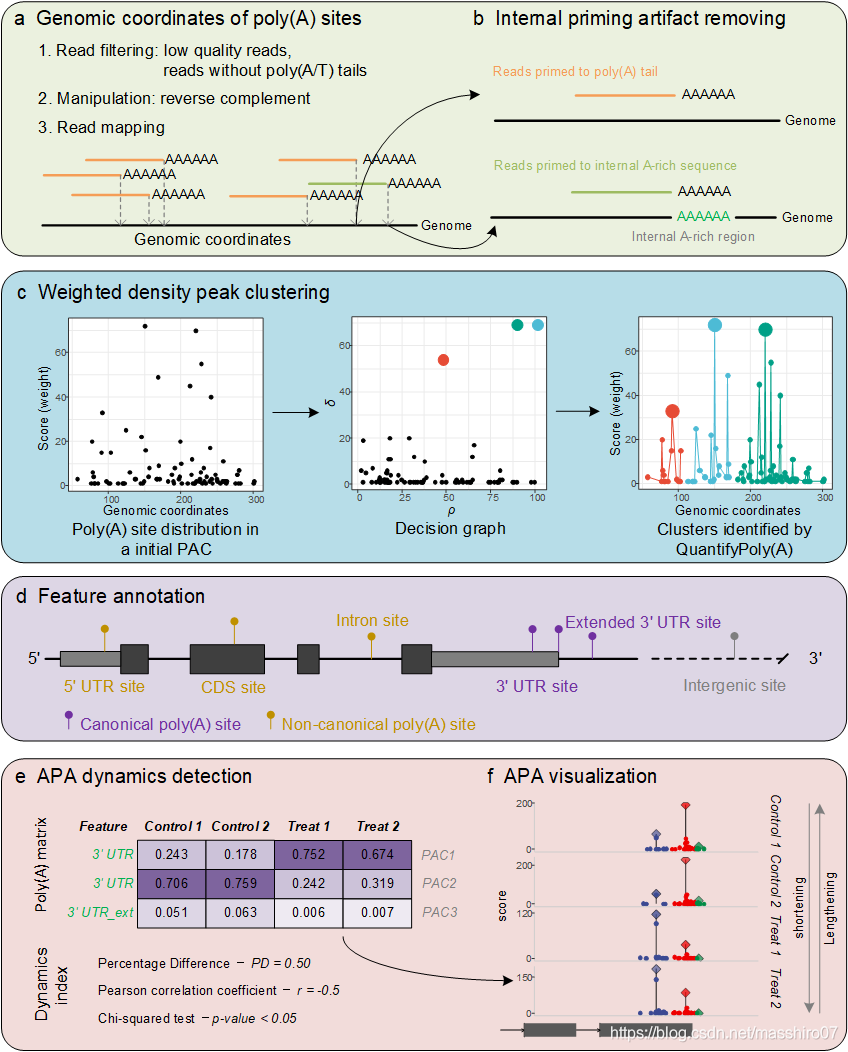
етИіRАќГіздРЯХЦЩњаХЦкПЏBriefings in BioinformaticsЩЯЕФЮФеТ QuantifyPoly(A): reshaping alternative polyadenylation landscapes of eukaryotes with weighted density peak clustering,СНЮЛЭЈбЖзїепЗжБ№ЮЊЯУУХДѓбЇЕФжњРэНЬЪквЖДгЭЅКЭИЃНЈХЉСжЕФИБНЬЪкСжПЁГЧ(дјдкЯУДѓзіВЉКѓ)ЁЃзїепжаЛЙгаРюЧьЫГРЯЪІ,РюРЯЪІЪЧжВЮяЖрЯймеЛЏбаОПЕФДѓФУЁЃДЫЧАЯУДѓвбОдкетИіСьгђЗЂБэСЫВЛЩйЮФеТ,АќРЈКмЖрбаОПаЭЮФеТЁЂвЛаЉЩњЮяаХЯЂбЇЗНЗЈвдМАжВЮяAPAЪ§ОнПтЕШ,ПЩвдЫЕвбОаЮГЩвЛЬзЭъећЕФPoly(A) site(PAS)ЪЙгУЕФЗжЮіСїГЬЁЃЕЋБОЮФвРШЛЪЧЖдЯШЧАЗНЗЈЕФвЛИіЭЛЦЦЁЃ
КмЖрНЈПтЗНЗЈЖМФмАяжњбаОПепЕЅМюЛљОЋЖШЕиЪЖБ№polyA siteЕФЮЛжУ,ЕЋЪЕМЪЩЯКЯВЂЯрСкЕФЮЛЕуГЩpoly(A) cluster(PAC)ЁЂПМТЧPACЕФЪЙгУВХИќКЯРэЁЃЫфШЛЮвУЧГЃЫЕpoly(A) Site,ЕЋетИіИХФюжЛдкЬжТлФГвЛДЮзЊТМЪТМўЪБгавтвхЁЃЕЋЪЕМЪЩЯ,МДЪЙЪЧЖдгкБрТыЭЌвЛЕААзЕФзЊТМБОЕФзЊТМЙ§ГЬ,ЖрЯймеЛЏВЂВЛвЛЖЈОЋШЗЕФЗЂЩњдкФГвЛИіМюЛљЩЯ,ЗЂЩњЕФЧјМфГЄЖШПЩФмдкМИИіМюЛљЕНЪЎМИИіМюЛљжЎМфЁЃЖдгкзЂЪЭНЯКУЕФФЃЪНЮяжж,етаЉЮЛЕуЭЈГЃОЭдкзЂЪЭЕФ3ЁЏЖЫИННќЁЃ(ДгетИіНЧЖШПДUTRЫЦКѕЪЧИіКмОЋЧЩЩшМЦ,вЛИіЛКГхЧј,зЊТМжежЙМДЪЙВЛФЧУДОЋШЗвВФмЗвыГіе§ШЗЕФЕААз)
Й§ШЅКЯВЂclusterвЛАуНіНівРППPASжЎМфЕФОрРы(БШШчОрРыдк24ИіМюЛљвдФкОЭКЯВЂ),вђЖјКЯВЂжЎКѓPACДѓаЁЛсЗжВМдквЛИіКмЙуЕФЗЖЮЇРя,БШШчЪ§ОнПтplantAPAdbжаЕФPACДѓаЁДг1ИіМюЛљЕНКУМИАйЕФЖМгаЁЃКмЖрДѓЕФPACУїЯдЪЧВЛКЯРэЕФЁЃдкетЦЊЮФеТжа,баОПепУЧОЭЬсГіСЫвЛжжаТЕФЫуЗЈ,ВЛНіЭЈЙ§ГЄЖШ,ЛЙвЊПМТЧУПИіЕуЩЯЕФread count,ИќзМШЗЕиШЅЖЈвхclusterЁЃ
ЕБШЛ,етЦЊЮФеТЕФвтвхВЛжЙгкДЫЁЃЛЙгавЛаЉЙигкЖЏжВЮяPACИННќmotifВювьЕФЗЂЯж,ЬсГіСЫЖЏЮяжаЕФБЃЪиЕФpolyA signal(AAUAAA)дкжВЮяжаПЩФмВЛЪЧзюживЊЕФаХКХ,ВЂЧвЗЂЯжСЫжВЮяPACЩЯгЮИЛМЏЕФUGUA КЭ UAAA motifЁЃ
НгЯТРДШУЮвУЧвЛЦ№ПДПДетИіАќЕНЕзШчКЮЪЙгУЁЃ
УћДЪЫѕаД
- Poly(A) Cluster ЈC PAC
- Poly(A) Site ЈC PAS
- alternative polyadenylation ЈC APA
ЖўЁЂАВзА
ЯТдиСДНгМАЯрЙивРРЕАќПЩвдВЮПМ ЙйЗННЬГЬ
ЭјеОеыЖдRАцБОВЛЭЌ(вдR 3.5.0ЮЊЗжНч,вђЮЊbioconductorЕФгУЗЈВЛЭЌ)ИјГіСЫСНЬзвРРЕАќАВзАДњТыЁЃ
ЪЕМЪАВзАЙ§ГЬжа,ШчЙћЪЧЕЅЖРАВзАвЛИіаТЕФRЛЗОГ,НЈвщАВзА3.6вдЩЯАцБО(БЪепгУСЫ3.6.3)ЁЃжЎЧАЯШГЂЪдгУ3.5.0АВзА,ЗЂЯжРЇФбжижи,ПЩФмвђЮЊАВзАЪБШчЙћВЛжИЖЈАќЕФАцБОвЛАуЛсЯТдиВЂАВзАзюаТАц,дкетИіRАќЕФвЛГЄДЎвРРЕАќжа,ДцдкзХ(вРРЕАќЕФ)вРРЕАќвРРЕИќИпАцБОЕФR(Р§ШчDEseq2ЕФвЛИівРРЕАќ latticeExtra),етЪБжБНгАДееНЬГЬАВзАОЭЛсзАВЛЩЯЁЃ3.6вдЩЯЕФRПЩвдБмУтетРрЮЪЬтЁЃ
ШчЙћдкcondaЛЗОГжаЕФRРяАВзА,АВзАВЛЩЯФГаЉАќЪБ,вВПЩвдГЂЪджБНггУcondaзА(conda install r-АќУћ),БШШчproj4ОЭПЩвдетбљзА:conda install -c conda-forge r-proj4ЁЃcondaАВзАЪБвВвЊзЂвтАќЕФАцБО,гаЪБзААќЛсгаВПЗжвРРЕБЛЬцДњГЩИќОЩЕФАцБО,дЫааЪБГіЯжВЛМцШнЕФЧщПіЁЃ
етИіАќАВзАЕФФбЕудкгквРРЕАќЖр,вРРЕАќЖМАВзАЭъОЭКмШнвзСЫЁЃзАЭъАќХмвЛЯТВтЪдЪ§ОнЁЃ
Ш§ЁЂЪЙгУ
ВтЪдЪ§ОнЕФдЫааЈCвдФтФЯНцЕФЪ§ОнЮЊР§
ВЮПМЙйЗННЬГЬ ЕкЫФНк:
4.1 Application of QuantifyPoly(A) in an Arabidopsis dataset
Ъ§ОнзМБИ
ВтЪдЪ§ОнРДдДгкTranscriptome Analyses of FY Mutants Reveal Its Role in mRNA Alternative Polyadenylation, ЗжЮіСЫжВЮяжаЕФ3ЁЏ МгЙЄвђзгFY(дкЮяжжМфБЃЪи,гыШЫЕФWDR33,НЭФИPfs2pЭЌдД)дкЪЖБ№polyAаХКХ(AAUAAAМАвЛаЉЯрЫЦађСа)жаЦ№ЕНЕФзїгУЁЃГ§ШЅfy-3,СэвЛИігУЕНЕФЭЛБфЬхЪЧoxt6,CPSF30ЕФЭЛБфЬх,вВЪЧдк3ЁЏЖЫМгЙЄжаЗЧГЃживЊЕФвЛИіЕААз,CPSF30КЭFYЕФЙиЯЕвВЪЧЮФеТЕФвЛИіжиЕуЁЃвђЮЊCPSF30КЭFYдкШЫРяЕФЭЌдДЕААзвбОБЛжЄУїдкpolyAаХКХЪЖБ№Й§ГЬжаЦ№зїгУСЫ,ЫфШЛжВЮяжаAAUAAAзїЮЊpoly(A)аХКХВЛШчЖЏЮяжаЕФБЃЪи(ЖЏЮяГЌЙ§50%,жВЮя10%зѓгвЕЋвбОЪЧеМБШзюДѓЕФСЫ),вРШЛПЩвддЄЦкетСНИіЭЛБфЬхжаЕФpolyAЮЛЕуЪЙгУЧщПігывАЩњаЭЛсгаБШНЯДѓЕФВювь,вВЛсгавЛаЉШЋаТЕФpolyAЮЛЕу,ЖдpolyAЮЛЕуЪЙгУЕФЗжЮіЯдЕУИёЭтживЊЁЃЮФеТЪЙгУЕФЪЧPAT-seqВтађЪ§Он,ЪЕМЪЩЯЮвУЧвВПЩвдЪЙгУЦфЫћИЛМЏpolyAЮВаХЯЂЕФНЈПтЗНЗЈБШШчPAS-seqЛђепDRSЕШ,НјаавЛаЉЩЯгЮЗжЮіКѓЕУЕНpolyAЮЛЕуЕФЮЛжУКЭЗсЖШаХЯЂВЂДцДЂдкbedЮФМўжаЁЃ
жЕЕУвЛЬсЕФЪЧ,ВтЪдЪ§ОнЕФbedЮФМўВЂВЛЪЧБъзМЕФbedЮФМў,гЩ4СаЪ§ОнзщГЩ,вРДЮЪЧChromosome,strand,start,read countЁЃШчЙћгУздМКЕФЪ§ОнашвЊЖдБъзМЕФbedНјаазЊЛЛЁЃ
дЫааВНжш
- МгдиЪ§Он:АбЫљгаbedЖМЗХдквЛИіЮФМўМаФк,гУ
dirжИЖЈЮФМўМаЮЛжУЖСШЁЫљгаЮФМў,вВПЩвджБНгжИЖЈвЊЖСШЁЕФЮФМўУћ, ЮФМўУћЕФУќУћИёЪНЪЧЁАбљБОУћ_жиИДЁБ,БШШчЁАwt_1ЁБ - ШЅГ§internal primingдьГЩЕФМйpolyA site: етИіВПЗжбигУРюЧьЫГзщвЛЙсЕФБъзМ,дкpolyAЮЛЕуЧАКѓ10bpЙВ20bpЕФЧјМфФк,ШчЙћгЩСЌај6ИіA(AAAAAA)Лђепдк10bpЕФЧјМфФкгаВЛЩйгк7ИіA,ОЭШЯЮЊЪЧinternel primingЁЃЖдгкЛљвђФкСЌајСљИіAПЩФмв§Ц№internal primingЮвБэЪОжЇГж,ЕЋЪЧЖдгкЗЧСЌајЕФAдкШШСІбЇЩЯЪЧЗёгаРћгкoligodTЕФНсКЯБэЪОДцвЩЁЃAnyway,ВтЪдЪ§ОнКмИЩОЛУЛгаетаЉ,ЮвздМКЕФЪ§Он(3ЁЏmRNA-seq)ДѓИХЛсШЅЕє10%,ИљОнНЈПтЕФЗНЗЈВЛЭЌПЩФмЛсгаВювь(БШШчжЎЧАгаЮФеТЫЕгУanchored oligodTПЩвдМѕЩйinternal priming)ЁЃ
- poly(A) siteОлРр:етИіАќзюКЫаФЕФЙІФмжЎвЛ,дкЦеЭЈЕФАДОрРы(default: 24 nt)ОлРрЕФЛљДЁЩЯдіМгСЫвЛВНРрЫЦpeak callingЕФВНжш,АбДѓЕФclusterгжЛЎЗжГЩаЁЕФsub-cluster,УПИіsub-clusterевЕНСЫвЛИіИпЕу(peak?)зїЮЊжааФЁЃ(аЇЙћдкПЩЪгЛЏВПЗжгаЬхЯж,етВПЗжЮвВЛШЗЖЈРэНтЕФЪЧЗёзМШЗ)
- зЂЪЭ:зЂЪЭЛљБОЪЧАДеезЂЪЭЮФМўНјааЕФ,ЕЋЖдгк3ЁЏUTRНјааЭиеЙ,АбЯТгЮСНБЖ3ЁЏUTRГЄЖШвдФкЕФЧјгђГЦЮЊext_3ЁЏUTR, дк3ЁЏUTRМАext_3ЁЏUTRЕФclusterБЛШЯЮЊЪЧОЕфЕФpolyAЮЛЕу,ЦфЫћЕФЕиЗНОЭЙщРрГЩЗЧОЕфЕФpolyAЮЛЕуЁЃБрТыЛљвђЕФexonЧјгђЛсЯИЗжГЩUTRКЭCDS,ЗЧБрТыЕФОЭЪЧexonЁЃ(ЮЪЬт:ЖдгкУЛгаUTRЧјгђЕФnon-coding gene,3ЁЏЯТгЮЧјгђжаЕФPASЪЧВЛЪЧОЭВЛПМТЧСЫ)
- Г§ШЅreadЪ§Й§ЩйЕФPAC:Г§ШЅдкЫљгабљБОжаread countЖМаЁгк10ЕФPAC,уажЕПЩвдИќИФ
- APAЖЏЬЌЕФСПЛЏ:ЪзЯШашвЊЗжзщ,ЩшжУзщУћ,жЎКѓПЩвдБШНЯзщМфВювьЁЃетИіАќЬсЙЉСЫКУМИжжБШНЯЗНЪН,БШНЯгагУгІИУЪЧQuantify.CNCAPA(БШНЯОЕфPACКЭЗЧОЕфPACЕФЪЙгУ),ДЫЭтЛЙгаQuantify.GeneAPA(БШНЯЛљвђМфЫљгаPACЪЙгУЕФВювь),Quantify.CanonicalAPA(БШНЯОЕфPACжЎМфЪЙгУЕФВюБ№)ЁЃЮФеТЖЈвхСЫвЛИіНаPercentage Difference(PD)ЕФСПРДКтСПPACЪЙгУЕФВювь, P D = ЁЦ i , j , k ЈO p i , k ? q j , k ЈO 2 ЁС m ЁС n \mathrm{PD}=\frac{\sum_{i,j,k}\left|{p}_{i,k}-{q}_{j,k}\right|}{2\times m\times n} PD=2ЁСmЁСnЁЦi,j,k?ЈOpi,k??qj,k?ЈO?ЛЙзіМЦЫуСЫpearsonЯрЙиЯЕЪ§,зіСЫchi-square testЛёЕУpжЕ(ШУЮвгаЕуВявьЕФЪЧетИіpжЕЪЧИіЦНОљжЕ)ЁЃPDжЕЕФБпНчКмШнвзРэНт,0ЪЧЭъШЋУЛБф,БШШчОЕфPACгыЗЧОЕфPACЪЙгУБШР§ЭъШЋУЛБф,1ЪЧБфЛЏМЋДѓ,БШШчЖдеежажЛгУОЕфPACЕЋЪЕбщзщЭъШЋВЛгУОЕфPACжЛгУвЛИіnovelЕФЗЧОЕфPACЁЃЕЋЪЧШчЙћЩњЮябЇжиИДдкФГаЉЛљвђЩЯжиИДадВЛКУ,жиИДМфБфЛЏЧїЪЦВЛвЛжТ,вВЛсЕУЕННЯДѓЕФPDжЕЁЃЕЋЮвВЂВЛЬЋРэНтФмВЛФмгУrЛђpШЅШЅГ§етаЉЛљвђЕФИЩШХЁЃДгетВНПЩвдЕУЕНвЛаЉPACЪЙгУгаВювьЕФЛљвђ,ВЂдкЯТвЛВНжшжаЛцЭМЁЃ
- poly(A) profileЕФПЩЪгЛЏ:СїГЬжаfЕФЭМ,АќКЌЛљвђНсЙЙКЭPACЕФЪЙгУаХЯЂ,ПЩвджБЙлЕФЗДгГбљБОМфЕФPACЪЙгУВювьЁЃВЛЭЌPACбеЩЋВЛЭЌ;ЖдвЛИіPAC,clusterЕФжааФЕуОЭЪЧАєАєЬЧЭМФЧИіАєЕФЮЛжУ,clusterРяЕФreadзмЪ§ОЭЪЧАєАєЬЧЕФИпЖШ,raw countЯдЪОГЩЕуЁЃ
- DESeq2ВювьЗжЮі:етИіРяУцЕФВювьЗжЮівтвхВЛЬЋДѓ,ЕЋЪЧПЩвдЛPCAЛђепUMAPЭМ,ПДЯТЩњЮябЇжиИДжиИДадКУВЛКУЁЂЛЙгабљБОМфВювьЕФДѓаЁЁЃ
МИИіаЁЮЪЬт
- НЬГЬЬсЙЉСДНгЖдгІЕФfasЮФМўШОЩЋЬхУћзж(ИёЪН:X)КЭbedЮФМўжаЕФИёЪН(ИёЪН:ChrX)ВЛЖдгІ,здМКИќИФЯТЛђепЪЙгУБ№ЕФTAIR10ЕФfastaЮФМўЖМПЩвд
- gtfЮФМўЯТдиСДНгЛљБОУЛЫйЖШ,ВЛжЊЕРЪЧВЛЪЧЮвздМКЕФЮЪЬт,вВПЩвдгУвбгаЕФTAIR10АцБОЕФЮФМўДњЬц
- етЬзЪ§ОнПЩФмвбОШЅГ§Й§internal priming,дЫааЕкЖўВНВЂУЛгаШЅГ§ШЮКЮвЩЫЦinternal priming artifacts,ЮвгУbedtoolsЬсШЁађСаМьВщPASИННќЕФађСа,ШЗЪЕУЛгаAAAAAA
ЫФЁЂзмНс
QuantifyPolyAФмАяжњЮвУЧЖЈвхPACВЂЧвБШНЯбљБОМфPACЪЙгУЕФВювьЁЃЕЋЪЧРяУцгавЛаЉНсЙћВЛЬЋзМШЗЁЃ