m6AmPred:��������������Ϣʶ��RNA N6,2��-O-��������(m6Am)λ��
����:2��
IF:3.864
����:https://www.ncbi.nlm.nih.gov/geo/
��վ:https://www.xjtlu.edu.cn/biologicalsciences/m6am
ǰ��
N6,2��-O-��������(m6Am)��һ�ֹ㷺�����ڸ���RNA�����ϵĿ������Ρ�m6Am������ѧ�����в����,����������о���ʾ������ϸ���������˵�Ӱ�졣��ȷʶ����Ǻ����ϵ�m6Amλ���������������ѧ����������Ҫ���������������m6AmPred,���ǵ�һ�����ڴӺ��Ǻ����һ�������е���ʶ��m6Amλ��������������m6AmPred�����ڴ���Dart�㷨�ļ����ݶ���ǿ(XgbDart)��EIIP-�������ձ��뷽���Ļ�����,��ͨ��10��������֤�Ͷ����������ݼ����в���ʱ,AuC����0.954,ʵ�������˹����Ԥ�����ܡ�Ϊ���ϸ���Ժ���֤m6AmPred������,��������������Դ��ʵ����֤��m6Amλ������˽�����֤��
һ������
�ڹ�ȥ�ļ�����,RNA�Ķ�̬�����Ŵ������Ѿ���Ϊ����ѧ�о���һ����Ҫ���㡣�Դӷ��ֵ�һ���ṹ���εĺ�������,�Ѿ��г���170��ת¼�����α������������������͵�RNA��������RNA����,����mRNA��rRNA��tRNA��snRNA,�����ڵ������﹦�ܷ�����ֳ��߶ȵ������Ժ�Ч��,������̥��ϸ���ķ�����ϸ���Ĵ���Ǩ�ƻ��ƶ���ѧ���ݱ���,��������Ǻ�������øͻ����������༲���ķ�չ��
N6,2��-O-��������(m6Am),һ����m7Gñ�����ڶ�������Ϸ��ֵ�����,��mRNA���ձ��ת¼������֮һ������б�����2��-O-������(Am)�ļ�����ȥ���������ǿ����,�漰��д�Ͳ������ס�һ��ñ�����Լ�ת��ø,���ữCĩ�˽ṹ��(CTD)-���������1 (PCIF1)�������m6Am,��������ص���(FTO)���ԡ�������Am�����Ρ����ܶ�m6Am������ѧ������֪֮����,��������о��Ѿ���ʼ��ʾm6Am����ǿmRNA�ȶ��Ժͷ���Ч���Լ��谭��ֱ������ϸ����������Ĺ��ܡ�m6Amλ���ȷ�����Բ���������ѧ����������Ҫ,���,һЩʪʵ����ʵ�鷽��,��MeRIP-seq��miCLIP-Seq��m6ACE-seq�ѱ��������ڼ���m6Amλ�㡣Ȼ��,m6Amλ���RNA������������������Ȼ�dz�����,��Ҫ����Ϊ����ʵ����ʵ�����������߳����Ŀ���ɱ��ߡ�ʵ��ʱ�䳤�������Ե͡��������о���,������ͼ����һ�ּ����������ʶ����Ǻ��������е�m6Amλ��,ʹ��һ�ּ����ݶ���ǿ�㷨,XgbDart��Ϊ��������
�Ѿ�������һЩ����������Ԥ����Ǻ�������Ŵ����εļ��㷽����Ȼ��,��������֪,��ĿǰΪֹ��û��m6Amվ��Ԥ�������á����,�������о���,�����������һ��m6AmԤ������m6AmPred,���������ھ�ȷʶ��RNA�����е�M6Amλ�㡣Ϊ�˱��ڷ������ǵ�Ԥ����,��������һ���û��Ѻõ����������,����https://www.xjtlu.edu.cn/biologicalsciences/m6am.�ṩ��Ԥ�����ǵ�m6Amվ��Ԥ����m6AmPred������õ���������ʵ����֤����,��ͨ���ṩ�ɿ��ļ��㷽��������m6Amվ���ĵ��о���Ȼ��,��������֪,��ĿǰΪֹ��û��m6Amվ��Ԥ�������á�
���,�������о���,�����������һ��m6AmԤ������m6AmPred,���������ھ�ȷʶ��RNA�����е�M6Amλ�㡣Ϊ�˱��ڷ������ǵ�Ԥ����,��������һ���û��Ѻõ����������,����https://www.xjtlu.edu.cn/biologicalsciences/m6am.�ṩ��Ԥ�����ǵ�m6Amվ��Ԥ����m6AmPred������õ���������ʵ����֤����,��ͨ���ṩ�ɿ��ļ��㷽��������m6Amվ���ĵ��о���
���������뷽��
2.1 ѵ�����Ͳ��Լ�
�������ݼ�(m6Amλ��)����������ĵ�������ֱ���m6Am���������л��,������ͨ��miCLIP-seq������HEK293ϸ��ϵ�ϲ��������ݴӻ�������ۺ����ݿ�(GEO)����,GEO��¼��ΪGSE63753��GSE78040����ǰ���о���ʾ����Ϊ41��������[nt]���м���һ��RNA����λ���RNA������ʾ����ѽ��,���������������������ʽ,��ͨ������41nt������������ǵ��������ݼ�,����ʵ�������Amλ��λ�����ġ�������m6Amλ�����ͬת¼���ϵ�δ����BCA���������ѡ��δ���ε�Amλ��(��������),������10���������ݼ���ͨ������10���������е�ÿһ������������������,��1:1���������ʹ�����10�����������ݼ����������ڼ�����ǵ�Ԥ�����ܽ���ƽ��,�Լ������β��졣
��������GEO���ݼ������ݱ����,Ȼ�����������ת¼�����ݼ��ͳ����RNA���ݼ�,�����������ת¼�����ݼ�����2447����������,�ڳ����RNA���ݼ�����1673���������С�������ת¼���ͳ���ĺ��Ǻ������ݼ�������ֳ�ѵ�����Ͳ��Լ�,����Ϊ8:2��Ȼ��,�������������ݼ��ֱ���Ϊѵ�����Ͳ��Լ�,����ģ�͵�³���ԡ���1�г���ÿ�����ݼ���վ��������
2.2 ������ȡ
����ѧϰ�㷨�ĸ߷��ྫ���ںܴ�̶�������������RNA����������ȡ�����б�����ԡ�Ϊ�˻���������,���о����������ֱ������,�������ữѧ����(NCP) +�������ܶ�(ND)�͵���-�����������(EIIP) +αEIIP(pseiip),�������ǵ����ܽ����˱Ƚϡ�
2.2.1.�����ữѧ���ʺͺ������ܶ�
�������ܶ�(ND)��ʾ��������ÿ��λ�õķֲ���Ƶ����Ϣ����Nλ��������ܶ�(di)����ͨ����(i + 1)λ֮ǰ���ֵ�N������(N)����i : di= n/i�����㡣���,�������С�AUAGUCAUAA��,A���ܶ��ڵ�1��3��7��9��10λ�ֱ�Ϊ1��0.67��0.43��0.44��0.50��
���Ƶ�,λ��2��5��8����UΪ0.50��0.40��0.38,λ��6����CΪ0.17,λ��4����GΪ0.25��
�����ữѧ���ʵı��뷽���ǻ����ĸ����Ǻ��������IJ�ͬ��ѧ�ṹ����Ƶġ����ֲ����������A.T.M. Golam Bari����[33]�����,����Ԥ��DNA�����еļ���λ��,�������㷺���ڱ���RNA������Ԥ��RNA����[34�C36]�����Ǻ�����ĸ���ɲ���,����(A)������(U)������(G)�������?���ݻ��ṹ������(��������A��G,һ������C��T)������(A��C)��ͪ��(G��T)�Ĵ��ڡ�ǿ(C��G)����(A��T)����Ĵ��ڶ���Ϊ���顣���ڸ���Ϣ�ͺ������ܶ���Ϣ,��������S(����Ϊl)�ĺ�����N�������������µ�ʽ�Ĺ�ʽNi ={xi,yi,zi,di }(i = 1,2,3,��l)��ʾ:
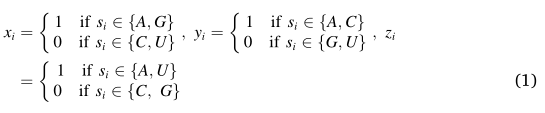
�����,A��C��G��U���Էֱ𱻱���Ϊ����(1,1,1,di),(0,1,0,di),(1,0,0,di)��(0,0,1,di)�����,һ�����Ǻ��������е�ÿ�������Ὣ��������ĸ���ֵ,ÿ�����Ǻ������н��������һ��4 *(�������еij���)-ά���塣
2.2.2.����-�����������(EIIP)��αEIIP
���ú�����ĵ���-�����������(EIIP)ֵ�ı��뷽����������ζ���˹���ɵ������,��������������������Ϣѧ�о�������EIIP,ÿһ����������ᶼ�������һ����ֵ,�������ĵ���-����������ơ���2������ÿ���������EIIPֵ��

�����ǵ��о���,EIIP��������һ������Ϊ41����������������,ͨ���������������ֵ�����ڸ��������е�Ƶ�����������αEIIP�����,�����κθ�����mRNA����,PseEIIP����һ��64ά����,������ʾ:
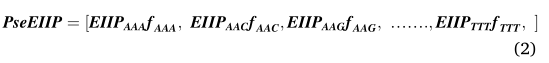
����,fxyzΪ���������Ƶ��,
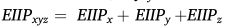
x,y,z����{ A,G,C,T}�������ǵ��о���,ͨ�����EIIP��PseEIIP,ÿ���������н���ת����41+64 = 105ά������
2.3 ��������
ʹ�����ָ�����������ǵ�ģ�͵�����:�����(������)�������(������)�������Ȼ��(����˹���ϵ��)�������Ȼ��(�ܾ���)�������Ȼ��(���ջ����������µ����),��ʽ����:

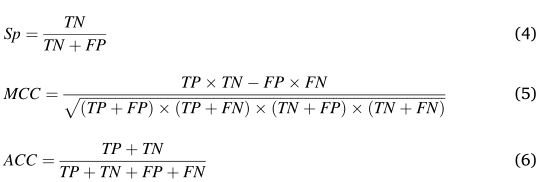
����TP��TN��FP��FN�ֱ���������ԡ������ԡ������ԡ������Ե�������Ӧ��0.5����ֵ���������������Ժ������ԡ�R��ggplot2���ڿ��ӻ�ģ�͵����ܡ�
2.4 ����ѧϰ��������ѡ��
֧��������(SVM)�����ɭ��(��Ƶ)������ģ��(GLM)��RNA����Ԥ���������еĻ���ѧϰ������,�ѱ��㷺���ڲ�ͬ������Ԥ�⡣����,�����ݶ���ǿ�㷨XgbDart (XGBDART)�ڱ��о��н����˲���,���㷨��ǰû�б�����RNA����Ԥ����ʹ�ù���XgbDart�������K. V. Rashmi��Ran Gilad-Bachrach�����,����ͨ����ϵ����ʹ��dropouts���˷�����ר�Ż������⡣����ͨ��10��������֤�Ͷ������Լ���������Щ�㷨�����ܡ�R������������ڹ�������ѧϰģ�͡�
2.5 ģ���Ż�
ͨ��ʹ�ò�ͬ���������г��Ⱥͳ���������,��һ���Ż�����ѡģ�͡����Ǻ�������(1)�ij������Ϊ2N+1,�м���һ��m6Amλ��,����������N (nt)���������С���3�г��˲�ͬ���ȡ���ͬ�������Ļ����Ǻ������С�

XgbDart�㷨�ij�����ͨ�������������������������ȡ�������(ѧϰ��)����С��ʧ���١��������ٷֱȡ��������бȡ��������ı��������������ĸ��ʺ�ʵ��Ȩ�ص���С����Ӧ������������������ѡ��������г��Ⱥͳ���������m6AmPred�����չ�����
2.6 ������Ҫ��
�˽����ѧϰԤ�ⱳ���ԭ���������������ѧϰ�㷨,�����������ģ�ͽ�������Ρ����,���Ƿ�������������Ҫ��,������������RNA���ݼ���ʶ��Ļ�������˱Ƚϡ�����RNA���еĻ���DREME�����ֹ��߲���
2.7 ����m6Am RNA���εĸ���
��m6Am�����������,���һ��վ���Ԥ��ֵ����0.5,���վ�㱻Ԥ��Ϊ�ٶ���m6Amվ�㡣Ȼ�����m6Amλ�����Ȼ��(LR)������m6Am RNA�����ĸ���,��СLRֵΪ1��LRֵ�ϴ��վ����������п�����m6Amվ�㡣

�������
3.1 ȷ��m6Amվ��Ԥ�����ѻ���ѧϰ�㷨��������ȡ����
���ַ�������ϲ�ͬ������ȡ�������������������ͼ1��ʾ��

ͼ1���������Լ����ĸ��������ֲ�ͬ���뷽���ķ�����������������ͼ1�Ƚ��˲�ͬ���뷽���IJ�ͬ��������AUCֵ����ͼ�Ƚ����ĸ���������ȫת¼ģʽ�����ֲ�ͬ���뷽����Ԥ������,��ͼ��ʾ�˳���RNAģʽ�µĽ����* XGBDART = XgbDart,SVM =֧��������,��Ƶ=���ɭ��,GLM =����ģ�͡�
���ڱ��뷽���ıȽ�,����EIIP-α������ȡʱ,���з����������ܶ�������NCP-ND����ķ����������ڷ������Ƚ�,XgbDart�����������㷨��ȡ������õ������������,����ѡ����XgbDart���EIIP-pseiip�����������������ѧϰģ�͡�
3.2 ģ���Ż�
ͨ�����������д�5nt����Ϊ60nt���Ż������ݼ��ij��ȡ����ܽϳ���������AUC��ȷ�Է�����ֳ��Ժõ�����,�����ǵ��������еij��Ƚ������Ƿ�������һ����ֵ,�û�����ϣ�������϶̵�����,����ѡ����40nt���н�һ���Ż���ģ�͵Ĵ��������ǵ���վ���ṩ,����û������ڱ���ѡ��ͬ�IJ�������(ͼ2)��

ͼ2��ͨ��AUC��ACC�Ż����г��ȡ�ͨ����m6Amλ����Χ���ò�ͬ�IJ�������,�Ż��˻����ݼ������г��ȡ�ģ�͵�����ͨ��AUC��ACCֵ���бȽϡ�
ͨ�����������Ż���XgbDartģ��,�����������ֵ���4��ʾ��

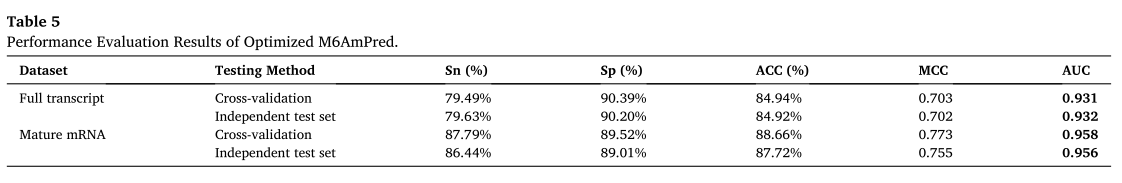
��5��ʾ��81nt�����ݼ����к��Ż���XgbDart���������������ܽ����
3.3 �����������ݼ�����������
ͨ��ʹ������������ͬ��Դ������,��һ������������ģ�͵��Ƚ��ԡ��ֱ�ʹ��GSE63753���ݼ���GSE78040���ݼ���Ϊѵ����,ͨ��10��������֤��������,Ȼ����һ�����ݼ���Ϊ�������Լ���������˵,��ʹ��GSE63753����ѵ��ʱ,��8:2�ı����������,����10��������֤,��ʹ������GSE78040���ж�������,��֮��Ȼ���������۽�����ڱ�6��

��ģ�;��нϸߵ�AUC,֤���˸�ģ�͵�³���Ժͷ���������
3.4 ��������
�ɲ�����Ű����ɵ�ǰ10����Ȩ������ͼ3d��ʾ��
����ģ�����������е���Ҫ�Զ�������������Ϊ����֤��Щ����,���ǻ������˳���RNA���ݼ��еĻ���,����ǰ3��������ͼ3a-c��ʾ��ֵ��ע�����,���������ɸ�����ʱ,�����ˡ�BCA������,�����������к����е�������,���Ԥ���߲�̫����ʶ�������еġ�BCA�������Ƚ�������������ʶ�����Ҫ�����ͻ���ʱ,���������ǵ�M6AmPred����ͨ��ʶ����CGG��GCG��CGC������Aλ��(N41)��Χ�ĺ��������������������������е�ģʽ��
ͼ3���������ĺ��Ǻ������к�������Ҫ�Է�����(a-c) DREME�Գ���RNA���еķ���ȷ������������:GCCAT��BCAB��GGCGGC��(M6AmPred��������Ҫ�Է�����ǰ10�����ܰ���Ҫ��˳�����С�
3.5 Webʵ��
�Ѿ�������һ�����������,�Ա��ڷ���M6AmPred��������https://www.xjtlu.edu.cn/biol�����ѧ/m6am���ɷ��ʡ��������û���FASTA��ʽ�ύ���ȳ���81nt�IJ�ѯRNA����,���������ṩ���չ�����������Ԥ������ʹ�����ǵ�M6AmPredԤ����Ԥ��m6Amվ��Ŀ����ԡ����мٶ���m6Amλ�㽫��ʶ��Ϊ������m6Amλ�����Ƶĺ��±��ʵ����,����Ԥ�������Ա��������ڽ�һ������(ͼ4��).

����ģ�͵Ĵ��������ǵ���վ���ز����ṩ,�������ǵ��û��ڱ�������ģ�͡�
�ġ�����
�ڱ��о���,������������������ͬ��Դ��m6Am����λ��,��������һ��XgbDartģ����Ԥ��m6Am����λ�㡣��ͨ�������IJ��Լ�����ʱ,���ǵ�ģ�ͻ�����൱�ߵ�ȷ��,��������ת¼��AUCΪ0.932,���ڳ���RNA AUCΪ0.956����ģ�ͻ�ͨ��ʹ������������Դ�����ݽ����˽���������
ֵ��ע�����,��Ϊһ��XgBoost�㷨,XgbDart������ʱĬ�ϻ�ռ�����е�CPU�ںˡ����ǵ�����ɱ�,���ǽ�web���������߳�����Ϊ4���ںˡ�����ģ�͵Ĵ��������ǵ���վ���ز����ṩ,�⽫�������ǵ��û������Ƶ��ڱ�����������
��ʹ��GSE63753��Ϊѵ����ʱ,10��������֤��AUC��ȷ�Գ������ϵص���ʹ������GSE78040��Ϊ�������Լ��Ľ���������ǵ�һ��֪ʶ��ͬ,��ģ��ͨ���Ȳ������ݼ����ʺ�ѵ�����ݼ���������Ϊ���������ܵ�ԭ������,GSE63753���ݼ����������ܽϲ�,���Ұ���һЩ������λ�㡣
Ȼ��,��ģ����Ȼ���Դ���ѧϰһЩģʽ,���,����ģ��Ӧ���ڸ��������ݼ�ʱ,�������ϣ������һ�ֿ��ܵĽ�����,GSE63753���ݼ���������������֮����һЩ�ض����������ݵ�,����ѧϰ�㷨���ѽ�����롣����������֮ǰ��Iris���ݼ��б��������и���������ݿ���ʱ,���ǽ��������ģ�͵��Ƚ��ԡ�
���о�������������������Ҫ���н�һ�����о�,ʹ�ö����ṹ����������Ϣ�����Ǻ������͵������������Ż�ģ��,�Խ�һ�����ģ�͵�³���Ժͷ���������