1. ǰ��
������Ӷ���ѧģ����õķ����DZ�дһ�����Ӷ���ѧ����,���仰˵,�̻�����ȥ���Ⲣ��ִ��,�Լ�����������ˡ���˱��Ľ��ӳ������������Ӽ�ķǼ�������е�Lennard-Jones potential����,�����Ӧ�õ�һ���ķ��Ӷ���������,��ģ���ά�ռ�������ķ��Ӷ���ѧ��Ϊ,Ϊ�˸���ֱ�۵������չʾ��һ����,������Ҫ��Python����ʵ����صĴ���ͳ���,��ʹ��jupyter notebook�����б���չʾ������,�ᾡ���ܵĽ��ͳ�ÿһ�д���ĺ���,�÷��Ӷ���ѧģ��ij�ѧ�ߺͰ������ܴ�0��1��д��һ�������ķ��Ӷ���ѧС����,�Լ���Է��Ӷ���ѧģ����̵����⡣
2. ���Ӽ��������
2.1 ���
�����ִ�������ѧ���ۿ�֪,���ı�����ʵ���������д��ڵ����������,���մ�ǿ������˳��,���ڵ�һ����ǿ�����,�����ʹԭ�Ӻ����������,�����ȶ��ش��ڡ����ڵڶ����ǵ�������,�������˻�ѧ��,���Ӽ��������,�Լ����ֺ������ĵ���,�����������,������,�����ȵȡ� ���ڵ��������������,��Ҳֻ����ԭ�Ӻ�����Ч,����ǿ�ȷdz���,��Ȼû��ʲô���ڸ�,���Ǻ���Ҫ,��������������������,��ǿ�ȼ���,�����ʵ�����û�пɹ۲��Ӱ��,���Ժ��Բ����ˡ�
��������Ҫ�����ɵ�����������ķ��Ӽ��������,��Ʒ��Ӽ�����,�ǽ鵼���Ӽ�����õ���,����������ԭ�Ӻ��������͵���������(����ԭ�ӻ�����)֮������������ų��������Ӽ��������(�Ǽ������) ������������������(�������) ��˵�����ġ�����,�漰ԭ��֮�乲�����ӶԵĹ��ۼ������ڷ���֮����ڵ���ǿ�öࡣ�����������Ƿ�����ѧ�г�����������Ҫ��ɲ��֡�
���Ӽ��������,����Ҫ����:
���»���(Van der Waals force):���Ϊ���������»����̶�������ձ�����ڹ̡�Һ����̬�κ���֮��,��������η��ɷ��ȡ�
�μ���(secondary bond): �������ڹ��ۼ������Ӽ���������,�����ڷ��»�����õ�����������á�
- ���(Hydrogen bonding):���뵪���������������������������
- �ǽ���ԭ�Ӽ�μ���:�����ڵⵥ�ʾ����С�
- ����ԭ����ǽ���ԭ�Ӽ�μ���:�����ڽ���������С�
- �����á�
- �������á�
�����ѧ��Ҳ�����о��µķ��Ӽ�������,����˫����ͽ���ȡ�
2.2 ���»���
���»���(Ӣ��:van der Waals�� Forces;����:�����߶�˹��)�ڻ�ѧ��ָ���ӻ�Ǽ���ԭ��(ϡ������)֮��Ƕ���ġ��ޱ����Եġ��������������,�����ų����������������ݺ�������ѧ��van der Waals(Լ����˹�������߶�˹)����,����1910���Noble�����������»����Ƿֱ��������������е�һ������ԭ��֮��ͨ��˲�侲������ò�����һ�����ķ���֮�����,�������ڸ������Ӳ�������������������(���Ӷ���ѧ�Ľṹ),�������,�����ܶȿ��ܻ���ʱ�����ת�Ƶ�ԭ�Ӻ˵�һ�ࡣ������һ��˲̬���,������ԭ�ӿ��Ա��������ų⡣�����Ȼ�ѧ���ۼ����ö�,ͨ��������С��5kJ/mol(0.4~4kJ/mol)��������ԭ��֮��ľ���Ϊ���ǵķ��»��뾶֮��ʱ,���»�������ǿ��ǿ�ķ��»��ų����ÿ��Է�ֹԭ������������»����Ĵ�С�ͷ��ӵĴ�С�����ȡ�
�ڽ��ܷ��»�������Դǰ,�ȼ��˽⼸������:
����ż��:���Է������������Ԫ�ز�ͬ,���������ӵ��������в���(Ԫ��������),���ʹ�÷������е���ƫ�Ƶ�����,�����Ͳ����˼���,����ż����������,��Ϊ����ż����
�յ�ż��:��ָ�Ǽ��Է����ڵ糡�л������������Է����ڽϽ�����������,���ڵ��Ӵ�����,�˴�����,���ǻᷢ��ƫ��,���������Ϊ�յ�ż����
˲ʱż��:һ�з�����,�����Ǽ��Է��ӻ��ǷǼ��Է���,ԭ�Ӻ�ʱ������,����ʱ�����˶���ԾǨ,�������˶���ʱ��,ż���뿪ƽ��λ�ö���������,ֻ����������̳���ʱ��ܶ�,�ʳ�˲ʱż����
���ݷ��»�������Դ�ɷ�Ϊ���¼�������:
1.ȡ����(1912-Keesom force):����ż��֮��ĵ�������;(��ƽ������ż����������ż�������)
���»������������ĵ�һ�������������ڹ���ż���ӡ��ļ���(���жԳ��Ե��������ķ���)�Ͷ༫��֮��ľ�������á�������ΪKeesom�����,��Willem Hendrik Keesom ��������������Щ��Դ�ڹ���ż����(ż������)֮���������,�������¶��йء�
Fig 1.ȡ����ʾ��ͼ
�����ɼ��ϵ�ż����֮����������������ɶ�ż���ӵIJ�ͬ��ת�������ƽ����������Ӳ�����ת������Զ���ᱻ������λ������һ���ܺõļ���,����ijЩʱ�����ȷʵ�ᱻ������λ��Keesom ����õ�����ȡ���ھ���ĵ������η�,�������ռ�̶�ż���ӵ����������ȡ���ھ���ĵ������η���Keesom �����ֻ�ܷ����ھ��й���ż���صķ���֮��,���������Է���֮����Keesom �����Ҳ�Ƿdz����ķ��»������,���ᷢ���ں��е���ʵ�ˮ��Һ�С����й���ż���� �� i \mu_{i} ��i?�� �� j \mu_{j} ��j?�ķ���i��j,�����������Ϊ:
E
e
=
?
2
3
[
��
i
2
��
j
2
(
4
��
��
0
)
2
K
B
T
1
r
6
]
(1)
E_e=-\frac{2}{3} \left [ \frac{\mu_{i}^2 \mu_{j}^2} {(4 \pi \varepsilon_{0})^2 K_B T} \frac {1}{r^6} \right ] \tag {1}
Ee?=?32?[(4����0?)2KB?T��i2?��j2??r61?](1)
E
e
E_e
Ee?��ʾ���ȡ���ż������õ�ƽ��������
�� 0 \varepsilon_{0} ��0? :���ɿռ�Ľ�糣��
T T T: �¶�
K B K_{B} KB?:������������,
r r r:���Ӽ���롣
2.�յ���(1921 Debye force):�յ�ż�������ż��֮��ĵ�������,�������յ�ż������ż���յ�ż������(��ƽ������ż�������յ�ż�������)
�ڶ�������Ϊ���Ϊ��Ӧ��(����)����°���,��Щ���Ժ�������������ѧ�ұ˵õ°ݵ�����������Ϊ���м����� �� \alpha ��???�ķ�������Χ���ӹ���ż���ص糡�������²����յ�ż����,�յ�ż��������Χ���ӹ���ż���ص�����á���һ�����й���ż���ӵķ����ų���һ�����ӵĵ���ʱ,�ͻᷢ����Щ�յ�ż���ӡ���������ż���ӵķ��ӿ��������Ƶ����ڷ������յ�ż���Ӳ������������ԭ��֮�䲻�ᷢ���°�������Ϊһ�㵥ԭ�Ӳ��Ƿ���,������,�����ԭ�ӷ�������������,Ϊ�Ǽ��Է�������ż��,����֮��û�������յ�,�ʵ°������������ԭ��֮�䡣�յ�ż���Ӻ���ż����֮��������� Keesom ����������������¶�,��Ϊ�յ�ż���ӿ���Χ�Ƽ��Է��������ƶ�����ת��������õ����ܱ�ʾΪ:
E p = ? �� i �� j 2 + �� j �� i 2 4 �� �� 0 1 r 6 (2) E_p=-\frac{\alpha_i \mu_j^2+\alpha_j \mu_i^2}{4\pi \varepsilon_{0}} \frac{1}{r^6} \tag {2} Ep?=?4����0?��i?��j2?+��j?��i2??r61?(2)
����������ż���ӵķ���(���� HCN)��û�з���ż���ӵķ�����ײʱ,��ײ������ͨ�������ڵ����ܶȵı仯����ż���ӳ��֡�HCN �еĵ�ԭ�Ӹ�������,����ż����ָ���ԭ�ӵķ�����ײʱ,�ڶ���ԭ�ӵĵ����ƻᱻ���϶���ĵ����ܶ��ų�,��˴������ԭ�Ӻ˻������ N ����������á�
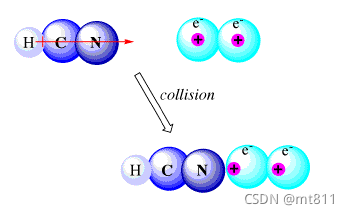
Fig 2.�յ�����ʾ��ͼ
3.ɫɢ��(London force):˲ʱż��֮��ĵ�������;(�յ�ż�������յ�ż�������)
������Ҳ����Ҫ�Ĺ�����ɫɢ������(����ż���յ�ż��),���ڷ����е��Ӻ�ԭ�Ӻ˲�ͣ���˶�,�Ǽ��Է��ӵĵ����Ƶķֲ��������������״̬,�Ӷ�ʹ����ԭ�Ӻ�֮�����˲ʱ���λ��,������˲ʱż��,����Ҳ����������Ρ������е��������ࡢԭ�������ࡢԭ�Ӱ뾶����,�������ױ���.˲ʱż����ʹ�����ڵ���һ�Ǽ��Է��Ӳ���˲ʱ�յ�ż��,������˲ʱż���ܲ�ȡ�켫����״̬,������ʱ�����ķ���˲ʱż�����������Ϊɫɢ��(���������ܱ���ʽ����ɫɢ��ʽ���ƶ�����)��˲��ż������ʱ�䲻�ϱ仯,ͳ��ƽ��Ϊ0,���ʵ������õ��ķ���ż��������ȻΪ0,ɫɢ����Ϊһ���������,����ʹ����ϵ��������,һ���,����������ֱ�Ϊ �� i \alpha_i ��i?�� �� j \alpha_j ��j?���������ӻ����Ӽ�ɫɢ�ܿ���д��:
E d = ? 3 2 [ I i I j I i + I j ] [ �� i �� j ( 4 �� �� 0 ) 2 1 r 6 ] (3) E_d=-\frac{3}{2} \left [ \frac{I_i I_j}{I_i+I_j} \right] \left [ \frac{\alpha_i \alpha_j}{(4 \pi \varepsilon_{0})^2} \frac{1}{r^6} \right] \tag {3} Ed?=?23?[Ii?+Ij?Ii?Ij??][(4����0?)2��i?��j??r61?](3)
��Ȼ˲ʱż�������ݶ�,���켫����״̬ȴ����˷�,�����ظ�,��˷��Ӽ�ʼ�մ�����ɫɢ��.����,ɫɢ�����������ڷǼ��Է��Ӽ�,Ҳ�����ڼ��Է��Ӽ��Լ�������Ǽ��Է��Ӽ䡣ɫɢ��������һ�з���֮��.ɫɢ������ӵı������й�,������ԽǿԽ�ױ�����,ɫɢ��ҲԽǿ��ϡ��������Ӽ䲢�����ɻ�ѧ��,����������ӽ�ʱ,����Һ�����ų�����,����ɫɢ�����ڵ�֤��.
���з���ͨ����ɫɢ�����յ�ż������á�����ͼ��,һ��2��ԭ�ӵķ�����һ��3��ԭ�ӵķ�����ײ����һ�����ӵĵ������ų���ײ���ķ��ӵĵ�����,����һЩ�����ܶ�Զ��ԭ�Ӻˡ�ԭ�Ӻ�Ȼ�����Լ��ĵ������εúܲ�,�������˵�һ�����ӵĵ����ơ�
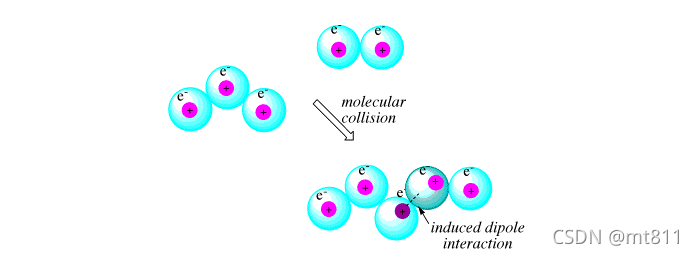
Fig 3.ɫɢ��ʾ��ͼ
4.������������ԭ���������ų�ɷ�,�ɷ�ֹ����̮����
�������۵ķ��Ӽ�����þ�Ϊ��������,������������Ӽ����ļ�С�����͡�����,��Щ��ϵ�����ʺϷ���������ر������������������������,�ﵽ��ӽ���������ص��ľ���ʱ,���Ӽ���ų����ý���������,������ϵ���ܵ�Ѹ�����ߡ�����,ԭ�Ӻ˼�ľ����ų�����,Ҳ������ϵ���������ߡ������ķ��Ӽ�����ú�������ͬʱ�����ų����õĹ��ס����۷�������, �ų����ö���ϵ�ƺ����Ĺ�������Ӽ�ľ����ָ����ϵ:
u
(
r
)
=
A
e
x
p
(
?
��
r
)
(4)
u(r)=A exp(-\beta r) \tag {4}
u(r)=Aexp(?��r)(4)
Ϊ�˼��㷽���Ч��,�����ų��ܱ�ʾΪ�ݺ�������ʽ:
u ( r ) = A / r n u(r)=A/r^n u(r)=A/rn
ʽ��,AΪ����;n����8~16�б仯��ͬʱ�ų���Ҳ��һ���߽�������á�����Ӽ���������Ѹ��˥������ԭ�Ӽ�������0.4 nm ʱ,��ʱ�ĶԷ��»����Ĺ�����Ҫ��������������ԭ��,�������ڲ������ų�,����Ϊ�ų�����

Fig 4. �����ӷ��»����������ķ������
����˵���»������ڿ��������(���������),�������Ǿ����������Ӽ�����ô����Ϊ����:��������úͷ��»��������(�����)��ǰ��˵���ķ��»������ھ��������,��Ϊ����ķ��»�����ͨ��ָ��ɫɢ��(London dispersion force),����������� E d E_d Ed?����Ӽ��(r)�� 1 r 6 \frac{1}{r^6} r61?�Ĺ�ϵ,������ķ��»���,����ΪKeesom force��Debye force����ƽ������London forceһ������ 1 r 6 \frac{1}{r^6} r61?�Ĺ�ϵ�����û����ƽ��,ż��-ż��������� 1 r 3 \frac{1}{r^3} r31?�Ĺ�ϵҲ����˵Keesom forceҪ���Ӷ̳�,����Զ̳�Ҳ�Ƿ��»���(�̳������)�����ھ��������(���������)����������˿��Կ����Ǽ�����÷�������Ϊ��������������仯��˥���ص㲻ͬ��
���»�������Ҫ�ص���:
-
���DZ���ͨ�Ĺ��ۼ������Ӽ�����
-
���»����ǿɼӵ�,���ܱ��͡�
-
����û�з������ԡ�
-
���Ƕ��Ƕ̳���,���ֻ��Ҫ�����������(��������������)֮�������á����Ӿ���Խ��,���»�����Խ��
-
����ż��-ż�������֮��,���»������¶��ء�
-
�ڵͷ���������,�伫���ǻ����������֧�������������ķ��»�����á��ڽϸ߷������Ĵ���,�Ǽ�������������ռ������λ�����������ǵ��ܽ�ȡ�
2.2.1 Lennard-Jones potential ģ��
Lennard-Jones potential,����John Lennard-Jones.(Լ�������ɵ�-��˹)��������������ΪLJ�ƻ���12-6�ơ��������˼�ԭ�Ӻͷ���֮������õĻ�������:��������õ������ڷdz����ľ�����ų�,���еȾ��������,���������ϲ������,LJ����һ����,�����Ʋ�����������������á�LJ�����о���㷺�������,����Ϊ�Ǽ���ʵ�ķ��Ӽ�����õİ뾭��ģ�͡�LJ�Ƶij��ñ���ʽΪ:
E L J = 4 �� [ ( �� r i j ) 12 ? ( �� r i j ) 6 ] (5) E_{LJ}=4 \varepsilon \left [ (\frac{\sigma}{r_{ij}})^{12} - (\frac{\sigma}{r_{ij}})^6 \right] \tag {5} ELJ?=4��[(rij?��?)12?(rij?��?)6](5)
E L J E_{LJ} ELJ?:Ϊ����֮������ܴ�С��
$\varepsilon $ :Ϊ��������õ�ǿ��(�� eV Ϊ��λ),�ӱ����Ͻ�,�����������������ǿ��,ͨ����Ϊ�������(Ҳ�С�ɫɢ�ܡ�);
�� \sigma ��:Ϊ����õ���������Ϊ��ʱ����ԭ�Ӽ�ľ���, �� \sigma �������������Ǽ������ӵĽӽ��̶ȵĶ���,��˱���Ϊ�����߶�˹�뾶�������ڷdzɼ�����֮��˼����Ķ���֮һ��
r i j r_{ij} rij?: Ϊi,j����֮��ľ��롣
��ʵ��Ӧ����, �� \varepsilon ��, �� \sigma ����������ͨ�������֪ʵ�����ݻ�ȷ���Ӽ�������ȷ���� ��һ��д����:
E L J ( r i j ) = ? [ ( r m i n r i j ) 12 ? 2 ( r m i n r i j ) 6 ] ( a m b e r �� �� �� �� ) (6) E_{LJ}(r_{ij})=\epsilon \left [ (\frac {r_{min}} {r_ij})^{12}-2(\frac {r_{min}}{r_{ij}})^6 \right ](amber�õ����) \tag {6} ELJ?(rij?)=?[(ri?jrmin??)12?2(rij?rmin??)6](amber��������)(6)
r m i n = 2 1 6 �� = 1.122 �� r_{min}=2^{\frac {1}{6}} \sigma=1.122 \sigma rmin?=261?��=1.122��:Ϊ����ײ�ʱ,�����ľ��롣
��������
�кܶ�����������»������,���������ΪLJ�ƺ�����Lennard-Jones ��ģ������������Ҫ��������ķ��������:�����ų���( 1 r i j 12 \frac{1}{r_{ij}^{12}} rij12?1?)����������������ӵĶ̾����������ص����ӹ������������������,������( 1 r i j 6 \frac{1}{r_{ij}^{6}} rij6?1?)�����˳����������(��ҪΪɫɢ��)��������,����������֮��������봦��ʧ���̾���Ķ����ų�����õ��¹����Һ��ĵ�ѹ����;���������ķ�ɢ����ö����������ȶ�����,��������Һƽ�⡣
������ĺ�����ʽ,��ָ����6��,���������ϵĺ�����,���ھ���ָ����12�����ų�����ϸ�ԭ�Ӻͷ���֮������������ɫɢ������Dz��ֵ�ɲ����Ľ�������ӻ�ѧ�������,����ɫɢ���ױ�����( 1 r i j 6 \frac{1}{r_{ij}^{6}} rij6?1?)���ٶ�˥����
( 1 r i j 12 \frac{1}{r_{ij}^{12}} rij12?1?) �ʹ��,��Ҫ����Ϊ���ڼ����Ͽ��Էdz���Ч��ʵ��Ϊ( 1 r i j 6 \frac{1}{r_{ij}^{6}} rij6?1?)��ƽ��,����ڡ�12�������ֵ������������,( 1 r i j 12 \frac{1}{r_{ij}^{12}} rij12?1?) �����ؽӽ�����������,����ʹ������ָ��������12��6���ƹ�Lennard-Jones����,���������ǽ�����LJ 6-12.
�����Ե�ԭ�ӷ����Ϊ��,Ӧ��LJ���ܺ��������-벼�������,ʹ������Python���붨����LJ�Ƶ�ÿ����ɲ��ֺ�����õ���������Ȼ���-벼�LJ���ܵı仯������һ��ͼ�ϡ� ? \epsilon ?�� �� \sigma ��?��ֵ�����-�������йص�ֵ��
#��Ƕ��ͼ
%matplotlib inline
import numpy as np
import matplotlib.pyplot as plt
#1.LJ���ܺ�����Ҫ����:...............................................................
#������������
def attractiveEnergy (r,epsilon,sigma):
return -4 * epsilon * np.power(sigma/r,6)
#�����ų���
def repulsiveEenergy(r,epsilon,sigma):
return 4 * epsilon * np.power(sigma/r,12)
#����LJ 6-12 ���ܺ���
def ljEenergy(r,epsilon,sigma):
return attractiveEnergy(r,epsilon,sigma) + repulsiveEenergy(r,epsilon,sigma)
#.....................................................................................
#2.ͼ�����....................................................
#���-�ԭ�Ӽ��LJ�����ͼ��(����:a=epsilon; b=sigma)
a=0.0103
b=3.4
#Ԥ���-�ԭ��Ϊ3��8��,��������100��ϵ������
r=np.linspace(3.0,10,100)
#�������������ͼ��
plt.plot(r,attractiveEnergy(r,a,b),label="Attractive")
#�����ų������ͼ��
plt.plot(r,repulsiveEenergy(r,a,b),label="Repulsiver")
#����
plt.plot(r, ljEenergy(r, a, b),
label='Lennard-Jones')
#������������
plt.xlabel(r'$r$(?)')
plt.ylabel(r'$E$(eV)')
plt.legend(frameon=False)
#ѡ��X,Yz��һЩ��ʾ��Χ,һ����չʾ��ԭ�Ӽ�����ñ仯����
plt.xlim([2.9, 8])
plt.ylim([-0.018, 0.034])
#����Ϊͼ���е�ע����Ϣ............................................................
#��(0,0)������ˮƽ����
plt.hlines(0, 0, 8, colors = "black", linestyles = "dashed")
# �����������espsilon,��ʱ��ԭ�Ӽ�����Ϊ:r=1.122*\sigma=3.8148
plt.annotate(s='$r_{min}=1.122 \sigma$', weight='heavy',xy=(3.8148, -0.0103), xytext=(5, -0.0103),
arrowprops=dict(arrowstyle='<-',))
plt.annotate(s='',xy=(3.8148, -0.0103), xytext=(3.8148, 0),
arrowprops=dict(arrowstyle='<->',color='red'))
plt.annotate(s='',xy=(2.91, 0), xytext=(3.4, 0),
arrowprops=dict(arrowstyle='<->',color='red'))
plt.text(3.9, -0.006, r'$\varepsilon$', fontsize=20)
plt.text(3, 0.003, r'$\sigma$', fontsize=20)
# ����ͼ��
#plt.show() # ����%matplotlib inline �Ϳ���ʡ��plt.show()��
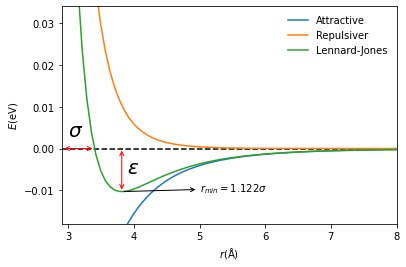
***Fig 5.����Python�����������ԭ�ӷ��Ӽ�LJ����������֮��仯�ĺ���ͼ��***
��ѧ��ʾ
�������ϵĵ�ʽ֮��,���м��ֲ�ͬ����ʽ����ʾLJ���ܺ�����
AB��:
AB��ʽ���������ڷ���������ʵ��,��Ϊ���������,���ʾ�ľ���LJ��
E L J = A r i j 1 2 ? B r i j 5 (7) E_{LJ}=\frac{A}{r_{ij}^12}-\frac{B}{r_{ij}^5} \tag {7} ELJ?=rij1?2A??rij5?B?(7)
����:
A = 4 �� �� 12 A=4 \varepsilon \sigma^{12} A=4����12
B = 4 �� �� 6 B=4 \varepsilon \sigma^6 B=4����6
�� = A B 6 \sigma=\sqrt[6]{\frac{A}{B}} ��=6BA??
�� = B 2 4 A \varepsilon=\frac{B^2}{4A} ��=4AB2?
LJ�� n-m��:
��LJ�ƺ�����,ָ��12��6�ֱ����ų�����Ӳ�ȵĺ������������÷�Χ�йء��Զ����л�������˵,�� ( 1 r i j 6 ) (\frac{1}{r_{ij}^6}) (rij6?1?)���������ƺ���,Ч������,������ ( 1 r i j 12 ) (\frac{1}{r_{ij}^{12}}) (rij12?1?)�����ųⲿ��,���ų���̫����,������ 1 r i j 9 \frac{1}{r_{ij}^{9}} rij9?1?�� 1 r i j 10 \frac{1}{r_{ij}^{10}} rij10?1?���ʺ�ʵ�ʷ��ӡ����ʱ�����ֻ�ǵ�������A��B(���� �� \varepsilon ���� �� \sigma ��?),�����ﵽ���õ�Ч��,��˿�����ͨʽ��ʾ�ƺ���:
E L J = �� i j n ? m ( n n m m ) 1 n ? m [ ( �� r i j ) n ? ( �� r i j ) m ] (8) E_{LJ}=\frac{\varepsilon_{ij}}{n-m}(\frac{n^n}{m^m})^{\frac{1}{n-m}} \left [ (\frac{\sigma}{r_{ij}})^n -(\frac{\sigma}{r_{ij}})^m \right] \tag {8} ELJ?=n?m��ij??(mmnn?)n?m1?[(rij?��?)n?(rij?��?)m](8)
��һ��Ϊԭ��ʵ���ų���,nԽ��ԭ��ʵԽӲ;�ڶ���Ϊ���������÷�Χ,mԽС�����������÷�ΧԽ��,��ʵ��Ӧ��ʱ,���õ�LJ 12-10�ƺ����������,LJ 12-3�ƺ������ƽ���ԭ�Ӽ��������,Ч���Ϻá�
�ڷ���ģ���е�Ӧ��
LJ�Ʋ������ڼ��㻯ѧ������������ѧ��������Ҫ,���Ҷ�����ʵ���ʵĽ�ģҲ����ˡ�ʹ��LJ�Ƽ�����ĺ������,һ����������ȼ����ʵ�ʵ�������кܺõ�һ����,��һ�����ƺ��� E i j E_{ij} Eij?�����ӻ�ѧ�Ľ�����൱��һ���ԡ�ͬ����,LJ�ƶ�Һ���еķ���������кܺõ�����,���Թ����еķ����������ֻ�д��µ�����������Ҫ�����ڶ���������ڹ�����������Ҫ����,��LJ���в�������Щ���á����,LJ�Ʊ��㷺��������������ѧ���������,���ڹ�������ѧ��ȴ����ʹ�á����������,������������������ͼ����������,���ڷ���ģ���н���ɫɢ���ų�����õ�ģ�͡����Զ�������ԭ�Ӻͼ����ر�ȷ������,��������ԭ�Ӻͷ��ӵij�����Ͷ̾���ķ�������ü���,��Ҳ�ܲ���һ���ܺõĽ���ֵ�����,LJ�ƾ������������ӷ���(��������ˮ)�ķ���ģ�͵Ĺ���ģ��(�����еķǼ������������)���������������ǻ���LJ��,����TraPPE����,OPLS����,��MolMod����,(�Է��������ĸ������ڱ������۷�Χ֮��)�������Ƚ��Ĺ�̬���Ͻ�ģ,ʹ���˸����ӵĶ�����(��EAM��)������LJ�ƻ�������ģ���-Һ�����ϵ���������,������������ѧ������
3. ���Ӷ���ѧģ�����
3.1 ���
���Ӷ���ѧ�����Ļ���˼���Ǹ��ݷ��ӵ����ܺ���(����),�õ�������ÿ��ԭ���ϵ���,����ţ�ٵڶ���������˶�����,�õ�ԭ�����������ϵ��˶��켣,�Ӷ��ﵽ����������Ŀ�ġ�
�Ӿ�����ѧ�Ƕȷ���,������ϵ����һ����з����ںͷ��Ӽ�����õ�ԭ����ɵ���ѧ��ϵ����ԭ�Ӻ˼�����ԭ�ӵ���Ҫ����,�����и�ԭ�ӿ��Խ��Ƶؿ���λ����Ӧԭ�Ӻ�λ�õ�һ���ʵ�,��˷�����ϵ���Խ���Ϊ������ѧ��ϵ:
�����ںͷ��Ӽ�������ͨ���ɷ�����ϵ�����ܺ���������(����),���ݾ�����ѧ,ϵͳ�е�������ԭ��i�ϵ���Ϊ���ܵ��ݶ�,����:
F ? i = ? �� i E = ? ( i ? ? ? x i + j ? ? ? y i + k ? ? ? z i ) E (9) \begin{aligned} \vec F_i &=-\bigtriangledown_{i} E \\ &= - \left (\vec i \frac{\partial}{\partial x_i} + \vec j \frac{\partial}{\partial y_i} + \vec k \frac{\partial}{\partial z_i} \right) E \end{aligned} \tag {9} Fi??=?��i?E=?(i?xi???+j??yi???+k?zi???)E?(9)
��ţ�ٶ���,�ɵõ�iԭ�ӵļ��ٶ�Ϊ:
a
?
i
=
F
i
m
i
(10)
\vec a_{i}=\frac{F_i}{m_i} \tag {10}
ai?=mi?Fi??(10)
��ţ�ٶ��ɷ���ʽ��ʱ�����,����Ԥ��iԭ�Ӿ���ʱ��t����ٶȺ�λ��
KaTeX parse error: No such environment: align* at position 8: \begin{?a?l?i?g?n?*?}? \begin{aligned��
ʽ�� r ? \vec r r�� v ? \vec v v�ֱ�Ϊ���ӵ�λ�����ٶ�,�ϱꡰ0����ʾΪ���������ij�ʼֵ��?
���Ӷ���ѧģ����������о�LJ���ܺͻ�á����ɵ�-��˹���ʡ� �����������ԡ�LJ��ͨ���Ƿ�չ����(�ر���������)���۵ı�ѡ��,Ҳ�Ƿ�չ�Ͳ��Լ��㷽���Լ��㷨��ѡ��,����ʵ�����ʵ����Ե�������ģ�� �� \varepsilon ���� �� \sigma ����,LJ�Ʊ�ɺ�ȷ�ص�����������(��������)��
ʹ�÷��Ӷ���ѧģ���о���LJ����ģ������ɵ��ԭ�ӷ�����ϵʱ,�������ϵΪ���������Ӽ��������ɵ���ѧ��ϵ(��������ʹ��LJ�ƹ���),���Ӽ������LJ���ܺ���������,���ݾ�����ѧ,������ϵ֮����������������LJ�Ƶ���,����:
F ( r i j ) = ? �� E L J = ? d E L J d r r ^ = 4 ? [ 12 �� 12 r i j 13 ? 6 �� 6 r i j 7 ] r ^ = 48 ? [ �� 12 r i j 13 ? 24 ? �� 6 r i j 7 ] (12) \begin{aligned} F(r_{ij})&=-\bigtriangledown E_{LJ} \\ &= - \frac {dE_{LJ}}{dr}\hat r \\ &=4 \epsilon \left [ 12 \frac{\sigma^{12}}{r_{ij}^{13}}- 6\frac{\sigma^{6}}{r_{ij}^{7}}\right ] \hat r \\ &=48 \epsilon \left [ \frac{\sigma^{12}}{r_{ij}^{13}}- 24 \epsilon \frac{\sigma^{6}}{r_{ij}^{7}}\right ] \end{aligned} \tag {12} F(rij?)?=?��ELJ?=?drdELJ??r^=4?[12rij13?��12??6rij7?��6?]r^=48?[rij13?��12??24?rij7?��6?]?(12)
��ĿǰΪֹ,�����Ѿ������˽��˷��Ӷ���ѧģ���LJ�ƵĻ���ԭ��,���������ҿ�ʼΪ��ά�ռ���ԭ�ӷ�����ϵģ�ⴴ��һ������ģ��С����,�������������ij�����ѭ���ٱ�Ҫ֪ʶԭ��,�ᾡ���ܵļ��Ա�������Ӷ���ѧģ���һЩ��Ҫ��������
�������·�ʽ����:
(1)����ָ�����������IJ���(��ʼ�¶�,������,�ܶ�,����(ʱ��),������)��
(2)��ϵ��ʼ��(��ʼ�����ӵ�λ��������ٶ�)��
(3)�������������������ϵ�����
(4)��ţ���˶����̡���һ������һ��������ģ��ĺ��ġ��ظ�������ֱ�����Ǽ�����ϵ���ݻ���ָ����ʱ�䳤�ȡ�
(5)����ѭ�����֮��,���㲢��ӡָ����ϵ����ѧ�ⶨ����ƽ��ֵ,����,ģ�������
������Ҫ��ʹ��python����ʵ��,��ȻPython�����C����ִ���ٶȽ���,���Ƕ�����ѧϰΪĿ�Ķ������Լ���ģ��ʵ��Ŀ�Ķ��Ա�д����,ʹ��Python���б�����ڳ�ѧ����˵��Ϊֱ��,����,������������,Ҳ��������jupyter notebook����������python IDE�������νӵı���Ͳ����Լ����С�
�ڿ�ʼ����ǰ,�����Ǽ����¸ó���Ļ���ģ��,����ͼ��ʾ:���ӻ���չʾ�˷��Ӷ�ѧģ����������е�,
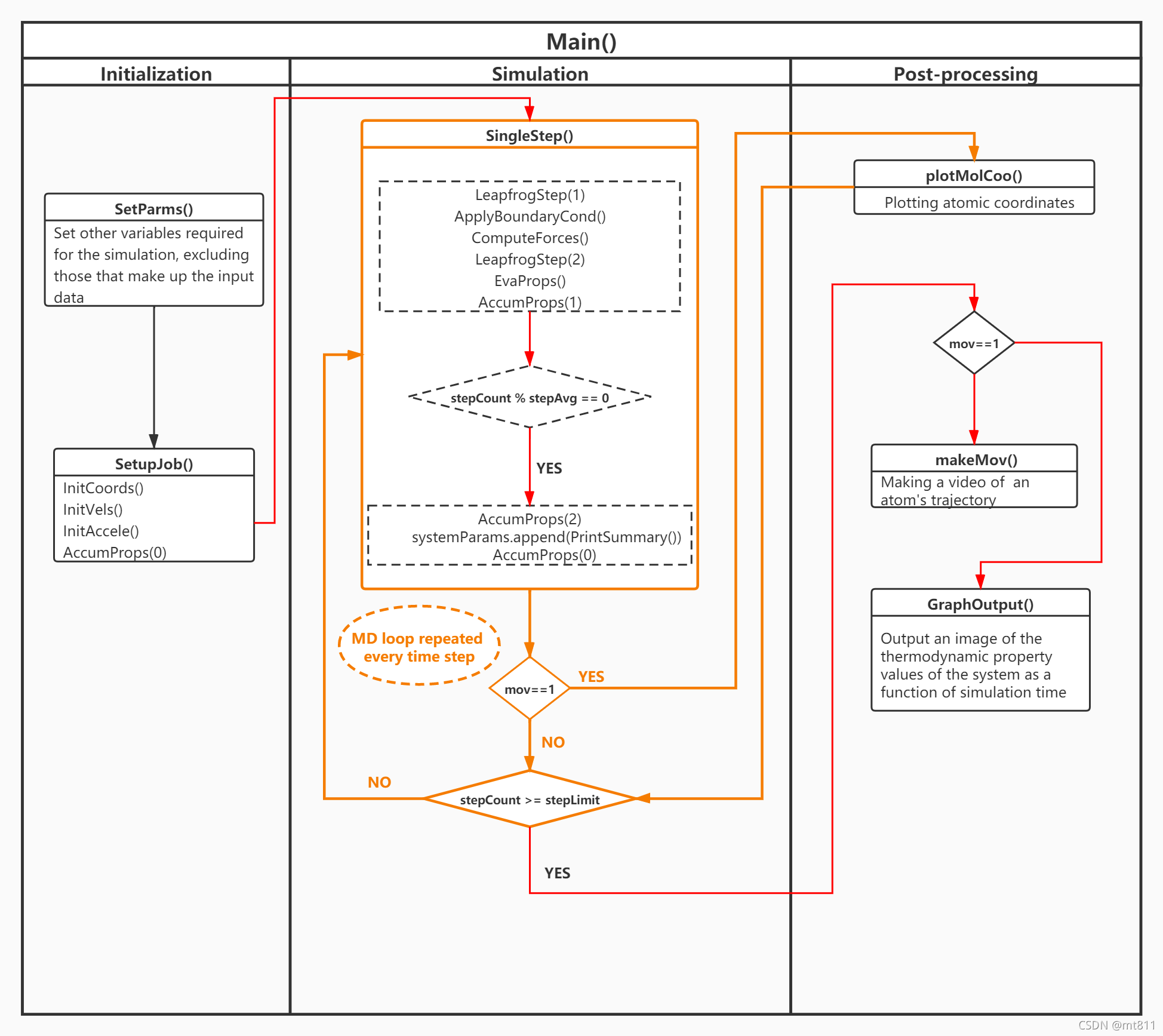
����ͼ����֪,��������(main)��Ҫ�����������:Initialzation(λ�ú��ٶȳ�ʼ��),Simulation(�������˶��ķ���),Post-processing(����,����ⶨ��)��
��Щ�������з��������һ�������Ĺ��ܺ���,�������ɰ����������ĺ���(���ڽ��µı�������н���)���ó�������еĹ�������:
����,����SetParams()��SetupJob()�������г�ʼ����SetParams()�������÷���ģ�������ȫ�ֱ���,��SetupJob()ִ��InitCoords(),InitVels()��InitAccels()�Ⱥ���,��ֱ��ʼ�����ӵ�����,�ٶȺͼ��ٶȡ�
���,����Simulation,�ù���Ϊ���Ӷ���ѧģ��ĺ��IJ���,����SingleStep()Ϊ��������ʱ�䲽���Ķ���ѧģ�����Ϊ�ĺ���,����Ҫ���ܾ��Ǹ������ӵ�λ�á��ٶȺ�������$ \Delta t $?ʱ��֮�����ӵ���λ�ú����ٶȡ������ȵ�����LeapFrogStep()��ִ���˶����̵Ļ���,��ͨ��ComputeForces()ʵ����LJ�ƺ����ļ���,�������ļ�����Ӧ���������Ա߽�����-ApplyBoundaryCond() ,���ִ�����������Բ����ĺ���:EvalProps()��AccumProps()������ִ��SingleStep()�ù��̵�ֱ��stepCount��������stepLimit�����ɽ�������Simulation���֡�
���,Post-processing,����plotMolCoo()�Ĺ��ܻ��Ƶ���ʱ�䲽����ģ����ϵ�����ӵ�λ�÷ֲ�����ͼ��,��������ԴΪSimuluation����ͨ����ţ���˶����̵õ����������µ����ꡣ��makeMov()Ϊ���ʱ�䲽���������˶��켣��Ƶ��GraphOutput()Ϊ���ģ����ϵ���Բ���ֵ(�ٶ�,������,����,ѹǿ�ľ�ֵ�ͱ���)�����ݺ�ͼ��
���濪ʼ����,����ÿһ�δ��붼�ᾡ���ܸ�����ϸ����,������Ҫ�����˽��ǰ��֪ʶ��������Ͳο����ϡ�
3.2 �����ļ�����
#�����ļ�(���Զ���):Rap_2_LJP.in
deltaT 0.005 #ʱ�䲽��
density 0.8 #�����ܶ�
initUcell_x 20 #�����x��С
initUcell_y 20 #y��С
stepAvg 100 #��������ͳ�Ʋ���
stepEquil 0 #ƽ�ⲽ��
stepLimit 500 #ģ��ѭ����
temperature 1. #�¶�����
3.3 ����1 :����ģ�������Ҫ�����������
import pandas as pd
import math
import os
import matplotlib.pyplot as plt
plt.style.use('seaborn-whitegrid')
import numpy as np
from PIL import Image
import glob
import moviepy.editor as mp
from datetime import datetime
import time
import os.path
from os import path
import shutil
from IPython.display import display
������
������������һ��ħ������(Magic Functions)���ٷ������Ķ�����:IPython��һ��Ԥ�ȶ���õ���ν��ħ������(Magic Functions),�����ͨ�������е����ʽ���������ǡ��ɼ���%matplotlib inline������ģ��������������magic��������IPython�ж��е���ʽ����Ȼ��IPython������magic����,��ô��Pycharm���Dz���֧�ֵ�,����juypter������Ч,ʹ��.py������������ע�͵���
������
#Ϊjupyter notebook����Ƕ����ͼ��,�ڴ���15��
%matplotlib inline
#Ϊjupyter notebook����ʾpd.DateFrame()��ʾ�����б�,�Ҵ���1
from IPython.display import display
3.4 ����2 :���������ϵ������Ķ���
# ������Ķ���
class Mol():
def __init__(self,r,rv,ra):
"""
ÿһ���Ӷ���(mol)�����е���������:
r:ԭ������
rv:ԭ���ٶ�
ra:ԭ�Ӽ��ٶ�����
"""
#��ʼ���������Ե�ֵ
self.r=np.asarray([0.0,0.0])
self.rv=np.asarray([0.0,0.0])
self.ra=np.asarray([0.0,0.0])
# ��ϵ������,��Ҫ��������ϵ����(�ⶨ��)��ƽ��ֵ�ͱ���
class Prop():
def __init__(self, val, sum1, sum2 ):
self.val=val #��ϵ�ⶨ����ֵ
self.sum1=sum1 #��ϵ�ⶨ����ֵ�ĺ�
self.sum2=sum2 #��ϵ�ⶨ����ֵ��ƽ����
3.5 ����3:���ߺ���
# ����ƽ��������ֵ�ĺ���:
def Sqr(x):
return (x * x)
def Cube(x):
return ((x) * (x) * (x))
# �������� (0,1) ��Χ�ڵľ��ȷֲ��������,��18.4
def RandR():
global randSeedP
randSeedP=(randSeedP * IMUL + IADD) & MASK
return (randSeedP*SCALE)
# �����ڶ�ά�ռ�������о��ȷֲ����������ĵ�λ����
def VRand(p):
s: float
s = 2. * math.pi * RandR()
p[0] = math.cos(s)
p[1] = math.sin(s)
return p
# �����Ա߽�(PBC)�㷨:
"""
��һ��������:
��ϵ�����ӵ������Լ��:
��ά��ϵ��,һ������Ԫ����������ͬ��Ԫ��(��Ϊ"����"),
��һ�����Ӵ��ұ߽߱紩��,��������߽߱��Ӧλ������ͬ���ӽ���,
�ɴ˱�֤��Ԫ����������Ŀ���������綯�����������غ㡣
��������½���:
region[0]Ϊx����Ԫ���ı߳�,��������x�������������Ϊ:[-0.5*region[0],0.5*region[0]]
regino[1]Ϊy�����Ԫ���ı߳�,��������x�������������Ϊ:[-0.5*region[1],0.5*region[1]]
��x����Ԫ��Ϊ��:����߳�Ϊ1x1��������Ԫ��
����������Ϊx>=0.5ʱ,Ϊ��ȷ����ǰ������һ��1x1Ԫ����,�й�ϵ:x-��λ��=1,
����:���=x-1
ͬ����: x<-0.5ʱ,�й�ϵ:��λ��-(x)=1 ����:��λ��=1+x,
�ڶ�����:�����Ӽ�������ʹ����С������,��:ComputerForces()����
"""
#PBC�㷨��һ����:����ϵ�����������Լ��
def VWrapAll(v):
#��Ԫ��x���������Լ��
if v[0]>=0.5*region[0]:
v[0]-=region[0]
elif v[0]<-0.5*region[0]:
v[0] +=region[0]
#��Ԫ��y���������Լ��
if v[1] >= 0.5 * region[1]:
v[1] -= region[1]
elif v[1] < -0.5 * region[1]:
v[1] += region[1]
3.6 ����4: main()������
mov = 1 # ���������һ����Ƶ�Ļ�,���� mov=1
workdir = str(os.getcwd()+'/') # Ϊ����png����Ƶ����һ������Ŀ¼
# ���/cooĿ¼������,�ͽ�����,�����ɾ��/coo(����������)
# ����һ���µ�/cooĿ¼��
if path.exists(str(workdir+'coo'))==False:
os.makedirs(str(workdir+'coo'))
else:
shutil.rmtree(str(workdir+'coo'))
os.makedirs(str(workdir+'coo'))
# ������������ļ�
df_params = pd.read_csv('Rap_2_LJP.in', sep='\t', header=None, names=['parameter', 'value'])
#��ʼ��ģ��ϵͳ����ѧ����ֵ
NDIM = 2 #��������
vSum = np.asarray([0.0, 0.0]) #�ٶ�֮��
kinEnergy =Prop(0.0, 0.0, 0.0) #����
totEnergy =Prop(0.0, 0.0, 0.0) #����
pressure =Prop(0.0, 0.0, 0.0) #ѹǿ
systemParams = [] #��ʼ��ϵͳ�����б�
#���ڲ���������㷨�����ı���
IADD = 453806245
IMUL = 314159269
MASK = 2147483647
SCALE = 0.4656612873e-9
randSeedP = 17
#��ʼ������:��Rap_2_LJP.�ļ��еIJ���ֵ���ݸ�MDģ������Ҫ����ر���
deltaT = float(df_params.values[0][1]) #ģ�ⲽ��
density = float(df_params.values[1][1]) #��ϵ�ܶ�
initUcell = np.asarray([0.0, 0.0]) #��ʼ��Ԫ��,�����С�������ļ��ܶȾ���
initUcell[0] = int(df_params.values[2][1]) #Ԫ��X����=20
initUcell[1] = int(df_params.values[3][1]) #Ԫ��X����=20
stepAvg = int(df_params.values[4][1]) #����ƽ����
stepEquil = float(df_params.values[5][1]) #ƽ�ⲽ��
stepLimit = float(df_params.values[6][1]) #���ƵIJ���
temperature = float(df_params.values[7][1]) #��ϵ�¶�
#������һ��Mol����������,��СΪ20*20=400. mol[0]~mol[399],��400���ԭ�ӷ���
mol = [Mol(np.asarray([0.0, 0.0]), \
np.asarray([0.0, 0.0]), \
np.asarray([0.0, 0.0])) for i in range(int(initUcell[0]*initUcell[1]))]
global nMol #������ӵ���������
nMol = len(mol) #Ϊmol����ij���Ϊ400
# �-�����õ�LJ�Ʋ���:
epsilon = 1
sigma = 1
# ִ����ϵ��ʼ����ع��ܺ���
SetParams() #�����������ڲ�ģ���������
SetupJob() #��ʼ�����ӵ�����,�ٶ�,����
moreCycles = 1 #MDѭ�����ƿ��ر���1:run; 0:stop
n = 0 #��¼����,������ÿһ�����������ͼ����
while moreCycles: #MDѭ��
SingleStep() #���ÿһ�����˶�����
if mov==1:
plotMolCoo(mol, workdir, n) #����������������������ͼ
n += 1
if stepCount >= stepLimit:
moreCycles = 0
#���ģ����������ɵIJ���
columns = ['timestep','timeNow', '$\Sigma v$', 'E', '$\sigma E$', 'Ek', '$\sigma Ek$', 'P_1', 'P_2']
df_systemParams = pd.DataFrame(systemParams, columns=columns)
#���Ʊ���,����jupyter notebookʹ��,��ӡ�������б�
display(df_systemParams)
if mov==1:
makeMov() # ������Ƶ
GraphOutput() #�����ϵ�ⶨ����ģ��ʱ��仯��ͼ��
3.7 Initilaztion ���ֵĹ���
3.7.1 ����5:steParms()����
"""
ģ���������������,��������Щ�����������ݵı���,
�ɺ���SetParams���á�
"""
def SetParams():
global rCut #�ضϾ���
global region #����
global velMag #�ٶȱ仯����
#�ض�ֵΪ����ײ�ʱ��ԭ�Ӽ����,��rmin=rCut=2^1/6 *sigma
rCut=math.pow(2.,1./6. * sigma)
#������Ԥ���ܶ�������,������������Ĵ�С
"""
��ά�ռ��µ�:
desity=������Ŀ/���,
ƽ����ϵģ��,�ܶȱ��ֲ���,������Ŀ��������Ҫ�����ܶ����������ӻ����Ĵ�С,����:
"""
region=np.multiply(1./math.sqrt(density),initUcell)
nMol=len(mol)
#�ٶȵı仯����ȡ�����¶ȵĸߵ�
velMag = math.sqrt(NDIM * (1. -1. /nMol) * temperature)
3.7.2 ����6:SetupJob()����
"""
���г�ʼ������Ĺ��������������µĺ�����
"""
def SetupJob():
global stepCount #��������
stepCount = 0 #��ʼ����������������ֵ
InitCoords() #��ʼ������
InitVels() #��ʼ���ٶ�
InitAccels() #��ʼ�������ٶ�
AccumProps(0) #��ʼ��ϵͳ����ֵ(������,����,ѹǿ)
����7 :InitCoords(),InitVels(),InitAccels()
������Ҫ�dz�ʼ����ϵ���ӵ�����,�ٶ�,���ٶȵĺ���
# ��ʼ�����ꡣ
# ����ʹ����һ���������ξ���(����ѡ�ȳ��ı�),
# ���,ÿ��Ԫ��ֻ����һ��ԭ��,ϵͳ��ԭ��Ϊ����,
def InitCoords():
c=np.asarray([0.0,0.0]) #��ʼ����ֵ
gap=np.divide(region,initUcell) #������������Ԫ���Ĵ�С
n=0 #�����Ӽ���
#��400��ԭ�ӷֱ�ŵ�Ԫ����(��ЩԪ����x,y������������region����)
for ny in range(0, int(initUcell[1])):
for nx in range(0, int(initUcell[0])):
c = np.asarray([nx+0.5, ny+0.5])
c = np.multiply(c, gap)
c = np.add(c, np.multiply(-0.5, region))
mol[n].r = c
n=n+1
# ��ʼ���ٶȡ�
# ��ʼ�ٶȱ�����Ϊ�̶�����(velMag),
# ��ȡ�����¶ȡ��ڷ���������ٶȷ����
# �����ٶȷ���,��ȷ�������Ǿ�ֹ��,��Ϊϵͳ�ڲ��ܾ�������������,�������ٶ�Ӧ�ñ��ֲ���,
# ����vRand��Ϊ���ȷֲ��ľ���λ��������Դ��
def InitVels():
global vSum
vSum=np.zeros(vSum.shape) #�����ض���С,��0����µ�����:[0,0],��������״
for n in range(nMol):
VRand(mol[n].rv) #�����������ٶ�����[x,y]
mol[n].rv=np.multiply(mol[n].rv,velMag) #�����¶�,�����µ��ٶ�
vSum=np.add(vSum,mol[n].rv) #���ĵ��ٶ�,ϵͳ���ٶȸ��ʵ��ٶȵ��ܺ�
#�����ȷ���,��ȷ�������Ǿ�ֹ��,����ȡ400��������,x,y�����ƽ���ٶ�1/400*Vsum+ÿһ��ԭ�ӵ��ٶ�
for n in range (nMol):
mol[n].rv=np.add(mol[n].rv,np.multiply((- 1.0 / nMol),vSum))
#��ʼ�����ٶȡ�
# ���ٶȱ���ʼ��Ϊ��[0,0]
def InitAccels():
for n in range(nMol):
mol[n].ra=np.zeros(mol[n].ra.shape)
����8:AccumProps()
#��ʼ��ֵ
def PropZero(v):
v.sum1 = v.sum2 = 0.
return v
#�����ƽ����
def PropAccum(v):
v.sum1 += v.val
v.sum2 += Sqr(v.val)
return v
#��ƽ��ֵ�ͱ���
def PropAvg(v, n):
v.sum1 /= n
v.sum2 = math.sqrt(max(v.sum2 / n - Sqr(v.sum1), 0.))
return v
# AccumProps:�ռ��ⶨ���Ľ��,������Ҫ�����ƽ��ֵ�ͱ�ƫ�
def AccumProps(icode):
if icode == 0: # 0:��ʼ��
PropZero(totEnergy)
PropZero(kinEnergy)
PropZero(pressure)
if icode == 1: # 1:���
PropAccum(totEnergy)
PropAccum(kinEnergy)
PropAccum(pressure)
if icode == 2: # 2:��ƽ��ֵ�ͱ���
PropAvg(totEnergy, stepAvg)
PropAvg(kinEnergy, stepAvg)
PropAvg(pressure, stepAvg)
3.8 Simulation ���ֹ���
�ò�����һЩ���ڽ����������ѧ��ϵ���˶����̵���ֵ���㷽��,����ͳ����ѧ�б�����ϵƽ��̬����ѧ����(����,ѹǿ,�¶ȵ�)��ʽ�ʹ�������,���ڡ�The Art of Molecular Dynamics Simulation���ڶ���,�н�Ϊ��ϸ��˵��,�����ڴ����оͲ������ˡ�
3.8.1 ����9:SingleStep()
def SingleStep():
global stepCount # ��������
global timeNow # ģ������ʱ��
stepCount +=1
timeNow = stepCount * deltaT #ģ�����е�ʱ��=����x����
LeapfrogStep(1) #����˶����̻���
ApplyBoundaryCond() #Ӧ�������Ա߽�����
ComputeForces() # ����ԭ���������
LeapfrogStep(2) # ������ٶȵĻ���
EvalProps() #����ϵͳ����ֵ(�ٶ�,,�ٶ�ƽ����,������,����,ѹ��)
AccumProps(1) #ϵͳ����ֵ���
#ÿһ�ٲ�ͳ��ϵͳ������ֵ(0,100,200,300,400,500),��������stepAvg��ֵ�����Զ���
if (stepCount % stepAvg == 0):
AccumProps(2) #��ϵͳ������ֵ��ƽ��ֵ�ͱ���
systemParams.append(PrintSummary()) #��������뵽 systemParams�б���
AccumProps(0) # ����ϵͳ����ֵ������һ�ε�ͳ��
����10:LeapfrogStep( )
"""
��ν���ַ������Ǹ������ӵ�λ��,�ٶȺ���������deltTʱ��֮�����λ�ú����ٶ��㷨��
�˶����̵Ļ���ʹ��һ�ּ���ֵ����:ԾǨ�����÷����������õ������غ�����,
LeapfrogStep������������ٶȡ���������־���������ԾǨ���̵���һ������Ҫִ�С�
vix(t + h/2) = vix(t) + (h/2)aix(t)
rix(t + h) = rix(t) + hvix (t + h/2)
"""
def LeapfrogStep(part):
if part == 1:
for n in range (nMol):
mol[n].rv=np.add(mol[n].rv,np.multiply(0.5 * deltaT,mol[n].ra))
mol[n].r=np.add(mol[n].r,np.multiply(deltaT,mol[n].rv))
else :
for n in range(nMol):
mol[n].rv=np.add(mol[n].rv,np.multiply(0.5 * deltaT,mol[n].ra))
����11:ApplyBoundaryCond()
# Ӧ�������Ա߽�,Լ����������λ��,����Ԫ���������㶨
def ApplyBoundaryCond():
for n in range(nMol):
VWrapAll(mol[n].r)
����12:ComputeForces()
"""
#����ԭ�Ӽ������������
ComputeForce������:�����������(ԭ��)��λ�ü������ÿһ�����ӵ�����,
���������������ü��ٶȡ�
�ú���ʵ����LJ_force,��������λ��ri��rj��ÿһ��ԭ��i��j�ļ��ٶȺ�����
rCut=�������ٽ�ֵ(rc),����:rCut=math.pow(2., 1./6.)
��r��rCut����ʱ,���½���0��
ţ�ٵ���������ζ��fji=-fij,����ÿ��ԭ�Ӷ�ֻ��Ҫ���һ�Ρ�
"""
def ComputeForces():
global virSum #���ڼ���ѹǿ���м����(fij*rij)��
global uSum #LJ���ܺ�
fcVal=0 # ԭ��j��ԭ��iʩ�ӵ���
rrCut=Sqr(rCut) # rCut:Rc,�ضϰ뾶��ƽ��
#��ʼ�����Ӽ��ٶ�
for n in range (nMol):
mol[n].ra=np.zeros(mol[n].ra.shape)
uSum=0. #��ʼ��LJ���ܺ�ֵ
virSum=0.
n=0
for j1 in range(nMol-1):
for j2 in range(j1+1,nMol):
# ʹDelta Rij��(RJ1-RJ2��ƽ��֮��)
dr=np.subtract(mol[j1].r,mol[j2].r) # dr����Rj1��Rj2֮���x,y�����ֵ
VWrapAll(dr) #Ӧ��PBCԼ��ԭ����Ԫ����,������dr[0],dr[1]
rr=(dr[0] * dr[0] + dr[1] * dr[1]) #��ԭ�Ӿ����ƽ��,���ﲢδ����ԭ�Ӽ����ľ���ֵ,û��Ҫ,��Ϊ��ضϰ뾶ֻ�DZȴ�С,ʡȥ��ƽ�����ļ���
r=np.sqrt(rr) #r��Ϊԭ�Ӽ�ľ���
# ������ʹ��ԭ�Ӿ����ƽ���� dr2 < Rc^2 ���ж���ԭ�Ӽ��������,
if(rr < rrCut):
"""
��ԭ����:������ΪLJ��β����������Ը���ϵģ������������,����Ϊ�˼���ȥ����
����ԭ�������´���:
rri = sigma / rr
rri3 = Cube(rri)
fcVal = 48. * rri3 * (rri3 - 0.5) * rri
"""
#����ʵ����������LJ���ܼ���,������β�������������,���Ժ����IJⶨ���ļ���������ԭ�Ĵ��ڲ���
fcVal = 48 * epsilon * np.power(sigma, 12) / np.power(r, 13) - 24 * epsilon * np.power(sigma, 6) / np.power(r, 7)
# ���¼��ٶ�
mol[j1].ra = np.add(mol[j1].ra, np.multiply(fcVal, dr))
mol[j2].ra = np.add(mol[j2].ra, np.multiply(-fcVal, dr))
#LJ���ܼ���
uSum += 4 * epsilon * np.power(sigma/r, 12)/r - np.power(sigma/r, 6)
virSum += fcVal * rr
����13:EvalProps()
# ������ϵ����ѧ����ֵ(�ⶨֵ)
def EvalProps():
global vSum
vvSum = 0. #ϵͳ�ٶ�ƽ�����ܺ�
vSum = np.zeros(vSum.shape) #�����ٶ�,Ϊϵͳ�ٶ�=���ʵ��ٶ��ܺ�,��ʼ��Ϊ[0,0]
global kinEnergy #����
global totEenergy #������
global pressure #ѹǿ
#��������ٶ�
for n in range(nMol):
vSum =np.add(vSum,mol[n].rv)
vv = (mol[n].rv[0] * mol[n].rv[0] + mol[n].rv[1] * mol[n].rv[1]) #xv^2+yv^2
vvSum += vv
# �ڶ�ά������ϵ��,����ѧ����ֵ����ֵ���㷽��(���ο��鼮�ڶ���)
kinEnergy.val = (0.5 * vvSum) / nMol #����ԭ�Ӷ���,nMol����ԭ����
totEnergy.val = kinEnergy.val + (uSum / nMol) #������:����ԭ�ӵĶ���+����ԭ�ӵ�����
pressure.val = density * (vvSum + virSum) / (nMol * NDIM) #��ϵѹǿ
����14:PrintSummary( )
# ��ӡ�����ϵ�IJⶨ��
def PrintSummary():
#��ӡ�ⶨ��,����С�������λ
print(stepCount, \
"{0:.4f}".format(timeNow), \
"{0:.4f}".format(vSum[0] / nMol) ,\
"{0:.4f}".format(totEnergy.sum1),\
"{0:.4f}".format(totEnergy.sum2), \
"{0:.4f}".format(kinEnergy.sum1), \
"{0:.4f}".format(kinEnergy.sum2),\
"{0:.4f}".format(pressure.sum1),\
"{0:.4f}".format(pressure.sum2))
#���ؾ�ȷ�IJⶨ��ֵ
return (stepCount, \
timeNow, \
(vSum[0] / nMol) ,\
totEnergy.sum1,\
totEnergy.sum2, \
kinEnergy.sum1, \
kinEnergy.sum2,\
pressure.sum1,\
pressure.sum2)
3.9 Post-processing ���ֹ���
3.9.1 ����15:plotMolCoo( )
# ������������ͼ
def plotMolCoo(mol,workdir,n):
import matplotlib.patches as mpatches
import matplotlib.pyplot as plt
Time=timeNow #ģ��ʱ��
Sigma_v = "{0:.4f}".format(vSum[0] / nMol)
E = "{0:.4f}".format(totEnergy.sum1)
Sigma_E = "{0:.4f}".format(totEnergy.sum2)
Ek = "{0:.4f}".format(kinEnergy.sum1)
Sigma_Ek = "{0:.4f}".format(kinEnergy.sum2)
P_1 = "{0:.4f}".format(pressure.sum1)
P_2 = "{0:.4f}".format(pressure.sum2)
#�ô�����ʹͼ����jupyter notebook��ֱ����ʾ,��.py�����в���Ҫ
%matplotlib inline
TileName = (workdir+'coo/'+str(n)+'.png') #�����n��ʾn��,�������������ɵ�ͼ������
x = [] #�½��б���������x������
y = [] #�½��б���������y������
# ����0-400��ԭ�ӵ�����,���뵽�б���ȥ
for n in range(len(mol)):
x.append(mol[n].r[0])
y.append(mol[n].r[1])
# ���400��ԭ��������λ�ý�Ϊ���е�������ԭ��mol[250]��mol[251],���ڹ۲�
mark_1 = int(len(mol)/2 + len(mol)/8)
mark_2 = int(len(mol)/2 + len(mol)/8 + 1)
# ����ԭ���������ͼ��
plt.plot(x, y, 'o', color='blue') #ÿ��ԭ�Ӿ�Ϊ��ɫԲ��
plt.plot(x[mark_1], y[mark_1], 'o', color='red') #���mark_1Ϊ��ɫ
plt.plot(x[mark_2], y[mark_2], 'o', color='cyan') #���mark_2Ϊ��ɫ
# ����ͼ��ı���
plt.title('timestep:'+"{0:.4f}".format(timeNow)+'; '+\
'$\Sigma v$:'+Sigma_v+'; '+\
'E:'+E+'; '+\
'$\sigma E$:'+Sigma_E+';\n'+\
'Ek:'+Ek+'; ' +\
'$\sigma Ek$:'+Sigma_Ek+'; '+\
'P.sum1:'+P_1+'; '+\
'P.sum2:'+P_2+'; ', loc='left')
plt.savefig(TileName, dpi=100) #����Ϊ����ͼΪ.pngͼƬ
3.9.2 ����16:makeMov ( )
# ���������˶��켣��Ƶ
def makeMov():
# ������.pngͼ��תΪgifͼ��Ƭ��
frames = []
imgs = sorted(glob.glob('coo/*.png'), key=os.path.getmtime)
for i in imgs:
temp = Image.open(i)
keep = temp.copy()
frames.append(keep)
temp.close()
# ɾ��ȫ����.pngͼ
for i in imgs:
os.remove(i)
# Ƭ�κϲ�����ΪGIF�ļ�,��Զѭ������
frames[0].save('coo/coordinates.gif', format='GIF',
append_images=frames[1:],
save_all=True,
duration=30, loop=0)
# ��GIFͼת����MP4��Ƶ�ļ�
clip = mp.VideoFileClip("coo/coordinates.gif")
clip.write_videofile("coo/"+"coordinates"+".mp4")
# ɾ��gifͼ
os.remove("coo/coordinates.gif")
3.9.3 ����17:GraphOutput()
#���ƺ�ϵͳ����֮��ص�ͼ��
def GraphOutput():
ax = \
df_systemParams.plot(x="timestep", y='$\Sigma v$', kind="line")
df_systemParams.plot(x="timestep", y='E', kind="line", ax=ax, color="C1")
df_systemParams.plot(x="timestep", y='$\sigma E$', kind="line", ax=ax, color="C2")
df_systemParams.plot(x="timestep", y='Ek', kind="line", ax=ax, color="C3")
df_systemParams.plot(x="timestep", y='$\sigma Ek$', kind="line", ax=ax, color="C4")
df_systemParams.plot(x="timestep", y='P_1', kind="line", ax=ax, color="C9")
df_systemParams.plot(x="timestep", y='P_2', kind="line", ax=ax, color="C9")
plt.savefig('plot.jpg', dpi=300)
3.10 ��������
# ����1:..........................................................................................
import pandas as pd
import math
import os
import matplotlib.pyplot as plt
plt.style.use('seaborn-whitegrid')
import numpy as np
from PIL import Image
import glob
import moviepy.editor as mp
from datetime import datetime
import time
import os.path
from os import path
import shutil
# ����2:..........................................................................................
class Mol():
def __init__(self,r,rv,ra):
"""
ÿһ���Ӷ���(mol)�����е���������:
r:ԭ������
rv:ԭ���ٶ�
ra:ԭ�Ӽ��ٶ�����
"""
#��ʼ���������Ե�ֵ
self.r=np.asarray([0.0,0.0])
self.rv=np.asarray([0.0,0.0])
self.ra=np.asarray([0.0,0.0])
# ��ϵ������,��Ҫ��������ϵ����(�ⶨ��)��ƽ��ֵ�ͱ���
class Prop():
def __init__(self, val, sum1, sum2 ):
self.val=val #��ϵ�ⶨ����ֵ
self.sum1=sum1 #��ϵ�ⶨ����ֵ�ĺ�
self.sum2=sum2 #��ϵ�ⶨ����ֵ��ƽ����
# ����3:..........................................................................................
def Sqr(x):
return (x * x)
def Cube(x):
return ((x) * (x) * (x))
# �������� (0,1) ��Χ�ڵľ��ȷֲ��������,��18.4
def RandR():
global randSeedP
randSeedP=(randSeedP * IMUL + IADD) & MASK
return (randSeedP*SCALE)
# �����ڶ�ά�ռ�������о��ȷֲ����������ĵ�λ����
def VRand(p):
s: float
s = 2. * math.pi * RandR()
p[0] = math.cos(s)
p[1] = math.sin(s)
return p
# �����Ա߽�(PBC)�㷨:
# PBC�㷨��һ����:����ϵ�����������Լ��
def VWrapAll(v):
#��Ԫ��x���������Լ��
if v[0]>=0.5*region[0]:
v[0]-=region[0]
elif v[0]<-0.5*region[0]:
v[0] +=region[0]
#��Ԫ��y���������Լ��
if v[1] >= 0.5 * region[1]:
v[1] -= region[1]
elif v[1] < -0.5 * region[1]:
v[1] += region[1]
# ����5:..........................................................................................
def SetParams():
global rCut #�ضϾ���
global region #����
global velMag #�ٶȷ���
#�ض�ֵΪ����ײ�ʱ��ԭ�Ӽ����,��rmin=rCut=2^1/6 *sigma
rCut=math.pow(2.,1./6. * sigma)
region=np.multiply(1./math.sqrt(density),initUcell)
nMol=len(mol)
#�ٶȵı仯����ȡ�����¶ȵĸߵ�
velMag = math.sqrt(NDIM * (1. -1. /nMol) * temperature)
# ����7:..........................................................................................
def InitCoords():
c=np.asarray([0.0,0.0]) #��ʼ����ֵ
gap=np.divide(region,initUcell) #������������Ԫ���Ĵ�С
n=0 #�����Ӽ���
#��400��ԭ�ӷֱ�ŵ�Ԫ����(��ЩԪ����x,y������������region����)
for ny in range(0, int(initUcell[1])):
for nx in range(0, int(initUcell[0])):
c = np.asarray([nx+0.5, ny+0.5])
c = np.multiply(c, gap)
c = np.add(c, np.multiply(-0.5, region))
mol[n].r = c
n=n+1
def InitVels():
global vSum
vSum=np.zeros(vSum.shape) #�����ض���С,��0����µ�����:[0,0],��������״
for n in range(nMol):
VRand(mol[n].rv) #�����������ٶ�����[x,y]
mol[n].rv=np.multiply(mol[n].rv,velMag) #�����¶�,�����µ��ٶ�
vSum=np.add(vSum,mol[n].rv) #���ĵ��ٶ�,ϵͳ���ٶȸ��ʵ��ٶȵ��ܺ�
#�����ȷ���,��ȷ�������Ǿ�ֹ��,����ȡ400��������,x,y�����ƽ���ٶ�1/400*Vsum+ÿһ��ԭ�ӵ��ٶ�
for n in range (nMol):
mol[n].rv=np.add(mol[n].rv,np.multiply((- 1.0 / nMol),vSum))
def InitAccels():
for n in range(nMol):
mol[n].ra=np.zeros(mol[n].ra.shape)
# ����8:..........................................................................................
def PropZero(v):
v.sum1 = v.sum2 = 0.
return v
def PropAccum(v):
v.sum1 += v.val
v.sum2 += Sqr(v.val)
return v
def PropAvg(v, n):
v.sum1 /= n
v.sum2 = math.sqrt(max(v.sum2 / n - Sqr(v.sum1), 0.))
return v
def AccumProps(icode):
if icode == 0: # 0:��ʼ��
PropZero(totEnergy)
PropZero(kinEnergy)
PropZero(pressure)
if icode == 1: # 1:���
PropAccum(totEnergy)
PropAccum(kinEnergy)
PropAccum(pressure)
if icode == 2: # 2:��ƽ��ֵ�ͱ���
PropAvg(totEnergy, stepAvg)
PropAvg(kinEnergy, stepAvg)
PropAvg(pressure, stepAvg)
# ����6:..........................................................................................
def SetupJob():
global stepCount #��������
stepCount = 0 #��ʼ����������������ֵ
InitCoords() #��ʼ������
InitVels() #��ʼ���ٶ�
InitAccels() #��ʼ�����ٶ�
AccumProps(0) #��ʼ��ϵͳ����ֵ(������,����,ѹǿ)
# ����10:.........................................................................................
def LeapfrogStep(part):
if part == 1:
for n in range (nMol):
mol[n].rv=np.add(mol[n].rv,np.multiply(0.5 * deltaT,mol[n].ra))
mol[n].r=np.add(mol[n].r,np.multiply(deltaT,mol[n].rv))
else :
for n in range(nMol):
mol[n].rv=np.add(mol[n].rv,np.multiply(0.5 * deltaT,mol[n].ra))
# ����11:...................................................................................
def ApplyBoundaryCond():
for n in range(nMol):
VWrapAll(mol[n].r)
# ����12:...................................................................................
def ComputeForces():
global virSum #���ڼ���ѹǿ���м����(fij*rij)��
global uSum #LJ���ܺ�
fcVal=0 # ԭ��j��ԭ��iʩ�ӵ���
rrCut=Sqr(rCut) # rCut:Rc,�ضϰ뾶��ƽ��
#��ʼ�����Ӽ��ٶ�
for n in range (nMol):
mol[n].ra=np.zeros(mol[n].ra.shape)
uSum=0. #��ʼ��LJ���ܺ�ֵ
virSum=0.
n=0
for j1 in range(nMol-1):
for j2 in range(j1+1,nMol):
# ʹDelta Rij��(RJ1-RJ2��ƽ��֮��)
dr=np.subtract(mol[j1].r,mol[j2].r) # dr����Rj1��Rj2֮���x,y�����ֵ
VWrapAll(dr) #Ӧ��PBCԼ��ԭ����Ԫ����,������dr[0],dr[1]
rr=(dr[0] * dr[0] + dr[1] * dr[1]) #��ԭ�Ӿ����ƽ��,���ﲢδ����ԭ�Ӽ����ľ���ֵ,û��Ҫ,��Ϊ��ضϰ뾶ֻ�DZȴ�С,ʡȥ��ƽ�����ļ���
r=np.sqrt(rr) #r��Ϊԭ�Ӽ�ľ���
# ������ʹ��ԭ�Ӿ����ƽ���� dr2 < Rc^2 ���ж���ԭ�Ӽ��������,
if(rr < rrCut):
fcVal = 48 * epsilon * np.power(sigma, 12) / np.power(r, 13) - 24 * epsilon * np.power(sigma, 6) / np.power(r, 7)
# ���¼��ٶ�
mol[j1].ra = np.add(mol[j1].ra, np.multiply(fcVal, dr))
mol[j2].ra = np.add(mol[j2].ra, np.multiply(-fcVal, dr))
#LJ���ܼ���
uSum += 4 * epsilon * np.power(sigma/r, 12)/r - np.power(sigma/r, 6)
virSum += fcVal * rr
# ����13:...................................................................................
def EvalProps():
global vSum
vvSum = 0. #ϵͳ�ٶ�ƽ�����ܺ�
vSum = np.zeros(vSum.shape) #�����ٶ�,Ϊϵͳ�ٶ�=���ʵ��ٶ��ܺ�,��ʼ��Ϊ[0,0]
global kinEnergy #����
global totEenergy #������
global pressure #ѹǿ
#��������ٶ�
for n in range(nMol):
vSum =np.add(vSum,mol[n].rv)
vv = (mol[n].rv[0] * mol[n].rv[0] + mol[n].rv[1] * mol[n].rv[1]) #xv^2+yv^2
vvSum += vv
# �ڶ�ά������ϵ��,����ѧ����ֵ����ֵ���㷽��
kinEnergy.val = (0.5 * vvSum) / nMol #����ԭ�Ӷ���,nMol����ԭ����
totEnergy.val = kinEnergy.val + (uSum / nMol) #������:����ԭ�ӵĶ���+����ԭ�ӵ�����
pressure.val = density * (vvSum + virSum) / (nMol * NDIM) #��ϵѹǿ
# ����14:...................................................................................
def PrintSummary():
print(stepCount, \
"{0:.4f}".format(timeNow), \
"{0:.4f}".format(vSum[0] / nMol) ,\
"{0:.4f}".format(totEnergy.sum1),\
"{0:.4f}".format(totEnergy.sum2), \
"{0:.4f}".format(kinEnergy.sum1), \
"{0:.4f}".format(kinEnergy.sum2),\
"{0:.4f}".format(pressure.sum1),\
"{0:.4f}".format(pressure.sum2))
return (stepCount, \
timeNow, \
(vSum[0] / nMol) ,\
totEnergy.sum1,\
totEnergy.sum2, \
kinEnergy.sum1, \
kinEnergy.sum2,\
pressure.sum1,\
pressure.sum2)
# ����9:------------------------------------------------------------------------------------------
def SingleStep():
global stepCount # ��������
global timeNow # ģ������ʱ��
stepCount +=1
timeNow = stepCount * deltaT #ģ�����е�ʱ��=����x����
LeapfrogStep(1) #����˶����̻���
ApplyBoundaryCond() #Ӧ�������Ա߽�����
ComputeForces() # ����ԭ���������
LeapfrogStep(2) # ������ٶȵĻ���
EvalProps() #����ϵͳ����ֵ(�ٶ�,,�ٶ�ƽ����,������,����,ѹ��)
AccumProps(1) #ϵͳ����ֵ���
#ÿһ�ٲ�ͳ��ϵͳ������ֵ(0,100,200,300,400,500),��������stepAvg��ֵ�����Զ���
if (stepCount % stepAvg == 0):
AccumProps(2) #��ϵͳ������ֵ��ƽ��ֵ�ͱ���
systemParams.append(PrintSummary()) #��������뵽 systemParams�б���
AccumProps(0) # ����ϵͳ����ֵ������һ�ε�ͳ��
# ����15:...................................................................................
def plotMolCoo(mol,workdir,n):
import matplotlib.patches as mpatches
import matplotlib.pyplot as plt
Time=timeNow #ģ��ʱ��
Sigma_v = "{0:.4f}".format(vSum[0] / nMol)
E = "{0:.4f}".format(totEnergy.sum1)
Sigma_E = "{0:.4f}".format(totEnergy.sum2)
Ek = "{0:.4f}".format(kinEnergy.sum1)
Sigma_Ek = "{0:.4f}".format(kinEnergy.sum2)
P_1 = "{0:.4f}".format(pressure.sum1)
P_2 = "{0:.4f}".format(pressure.sum2)
%matplotlib inline
TileName = (workdir+'coo/'+str(n)+'.png') #�����n��ʾn��,�������������ɵ�ͼ������
x = [] #�½��б���������x������
y = [] #�½��б���������y������
# ����0-400��ԭ�ӵ�����,���뵽�б���ȥ
for n in range(len(mol)):
x.append(mol[n].r[0])
y.append(mol[n].r[1])
# ���400��ԭ��������λ�ý�Ϊ���е�������ԭ��mol[250]��mol[251],���ڹ۲�
mark_1 = int(len(mol)/2 + len(mol)/8)
mark_2 = int(len(mol)/2 + len(mol)/8 + 1)
# ����ԭ���������ͼ��
plt.plot(x, y, 'o', color='blue') #ÿ��ԭ�Ӿ�Ϊ��ɫԲ��
plt.plot(x[mark_1], y[mark_1], 'o', color='red') #���mark_1Ϊ��ɫ
plt.plot(x[mark_2], y[mark_2], 'o', color='cyan') #���mark_2Ϊ��ɫ
# ����ͼ��ı���
plt.title('timestep:'+"{0:.4f}".format(timeNow)+'; '+\
'$\Sigma v$:'+Sigma_v+'; '+\
'E:'+E+'; '+\
'$\sigma E$:'+Sigma_E+';\n'+\
'Ek:'+Ek+'; ' +\
'$\sigma Ek$:'+Sigma_Ek+'; '+\
'P.sum1:'+P_1+'; '+\
'P.sum2:'+P_2+'; ', loc='left')
plt.savefig(TileName, dpi=100) #����Ϊ����ͼΪ.pngͼƬ
# ����16:...................................................................................
def makeMov():
# ������.pngͼ��תΪgifͼ��Ƭ��
frames = []
imgs = sorted(glob.glob('coo/*.png'), key=os.path.getmtime)
for i in imgs:
temp = Image.open(i)
keep = temp.copy()
frames.append(keep)
temp.close()
# ɾ��ȫ����.pngͼ
for i in imgs:
os.remove(i)
# Ƭ�κϲ�����ΪGIF�ļ�,��Զѭ������
frames[0].save('coo/coordinates.gif', format='GIF',
append_images=frames[1:],
save_all=True,
duration=30, loop=0)
# ��GIFͼת����MP4��Ƶ�ļ�
clip = mp.VideoFileClip("coo/coordinates.gif")
clip.write_videofile("coo/"+"coordinates"+".mp4")
# ɾ��gifͼ
os.remove("coo/coordinates.gif")
# ����17:...................................................................................
def GraphOutput():
ax = \
df_systemParams.plot(x="timestep", y='$\Sigma v$', kind="line")
df_systemParams.plot(x="timestep", y='E', kind="line", ax=ax, color="C1")
df_systemParams.plot(x="timestep", y='$\sigma E$', kind="line", ax=ax, color="C2")
df_systemParams.plot(x="timestep", y='Ek', kind="line", ax=ax, color="C3")
df_systemParams.plot(x="timestep", y='$\sigma Ek$', kind="line", ax=ax, color="C4")
df_systemParams.plot(x="timestep", y='P_1', kind="line", ax=ax, color="C9")
df_systemParams.plot(x="timestep", y='P_2', kind="line", ax=ax, color="C9")
plt.savefig('plot.jpg', dpi=300)
# ����3:************************************Main************************************************
mov = 1 # ���������һ����Ƶ�Ļ�,���� mov=1
workdir = str(os.getcwd()+'/') # Ϊ����png����Ƶ����һ������Ŀ¼
if path.exists(str(workdir+'coo'))==False:
os.makedirs(str(workdir+'coo'))
else:
shutil.rmtree(str(workdir+'coo'))
os.makedirs(str(workdir+'coo'))
# ������������ļ�
df_params = pd.read_csv('Rap_2_LJP.in', sep='\t', header=None, names=['parameter', 'value'])
#��ʼ��ģ��ϵͳ����ѧ����ֵ
NDIM = 2 #��������
vSum = np.asarray([0.0, 0.0]) #�ٶ�֮��
kinEnergy =Prop(0.0, 0.0, 0.0) #����
totEnergy =Prop(0.0, 0.0, 0.0) #����
pressure =Prop(0.0, 0.0, 0.0) #ѹǿ
systemParams = [] #��ʼ��ϵͳ�����б�
#���ڲ���������㷨�����ı���
IADD = 453806245
IMUL = 314159269
MASK = 2147483647
SCALE = 0.4656612873e-9
randSeedP = 17
#��ʼ������:��Rap_2_LJP.�ļ��еIJ���ֵ���ݸ�MDģ������Ҫ����ر���
deltaT = float(df_params.values[0][1]) #ģ�ⲽ��
density = float(df_params.values[1][1]) #��ϵ�ܶ�
initUcell = np.asarray([0.0, 0.0]) #��ʼ��Ԫ��,�����С�������ļ��ܶȾ���
initUcell[0] = int(df_params.values[2][1]) #Ԫ��X����=20
initUcell[1] = int(df_params.values[3][1]) #Ԫ��X����=20
stepAvg = int(df_params.values[4][1]) #����ƽ����
stepEquil = float(df_params.values[5][1]) #ƽ�ⲽ��
stepLimit = float(df_params.values[6][1]) #���ƵIJ���
temperature = float(df_params.values[7][1]) #��ϵ�¶�
#������һ��Mol����������,��СΪ20*20=400. mol[0]~mol[399],��400���ԭ�ӷ���
mol = [Mol(np.asarray([0.0, 0.0]), \
np.asarray([0.0, 0.0]), \
np.asarray([0.0, 0.0])) for i in range(int(initUcell[0]*initUcell[1]))]
global nMol #������ӵ���������
nMol = len(mol) #Ϊmol����ij���Ϊ400
# �-�����õ�LJ�Ʋ���:
epsilon = 1
sigma = 1
# ִ����ϵ��ʼ����ع��ܺ���
SetParams() #�����������ڲ�ģ���������
SetupJob() #��ʼ�����ӵ�����,�ٶ�,����
moreCycles = 1 #MDѭ�����ƿ��ر���1:run; 0:stop
n = 0 #��¼����,������ÿһ�����������ͼ����
while moreCycles: #MDѭ��
SingleStep() #���ÿһ�����˶�����
if mov==1:
plotMolCoo(mol, workdir, n) #����������������������ͼ
n += 1
if stepCount >= stepLimit:
moreCycles = 0
#���ģ����������ɵIJ���
columns = ['timestep','timeNow', '$\Sigma v$', 'E', '$\sigma E$', 'Ek', '$\sigma Ek$', 'P_1', 'P_2']
df_systemParams = pd.DataFrame(systemParams, columns=columns)
#���Ʊ���,����jupyter notebookʹ��,��ӡ�������б�
display(df_systemParams)
if mov==1:
makeMov() # ������Ƶ
GraphOutput() #�����ϵ�ⶨ����ģ��ʱ��仯��ͼ��
3.11 ���г�����벽��:
���¹��̾��������������м��,���ﲻ����
- ��װpython 3
- ��װ�������������������(�ڴ���1���Ѿ�����,�Ѵ��ڵ����谲װ,���Լ�������ع��ھ�����Դ����װ����)
- �������ļ��������г���:
(��ѡ)��װװjupyter notebook��������,Ҳ�����ڱ���������(shell��dos)������
������ configura :���и÷��Ӷ���ѧģ�������ļ�
�� ������ Rap_2_LJP.in :����ģ����Ҫ����IJ����ļ�
�� ������ MD_LJP.py :��shell��dos)�����еij���
�� ������ MD_LJP.ipynb :jupyter notebook�����еij���
- ��ʼ����
- ��ʽ1:��������������,��������
python MD_ljp.py
- ��ʽ2:jupyter notebook������
��������:- (1).��MD_LJP.ipynb �����ļ���������jupyter notebook
- (2).��MD_LJP.ipynb�ļ�,��������С���ť����
- ���:
����:�� configura�ļ���������ģ�����,�������������ļ�:(�����̴������������,�ҵĵ������ýϲ���)
������ configura
�� ������ coo :����ģ��켣��Ƶ���ļ��� (����)
������coordinates.mp4 :MP4��ʽ��ģ��켣��Ƶ (����)
�� ������ plot.png :ģ����ϵ�ⶨ����ģ��ʱ��仯��ͼ��(����)
�� ������ Rap_2_LJP.in :����ģ����Ҫ����IJ����ļ�
�� ������ MD_LJP.py :��shell��dos)�����еij���
�� ������ MD_LJP.ipynb :jupyter notebook�����еij���
��jupyter notebook�������������,��ͼ��ʾ:
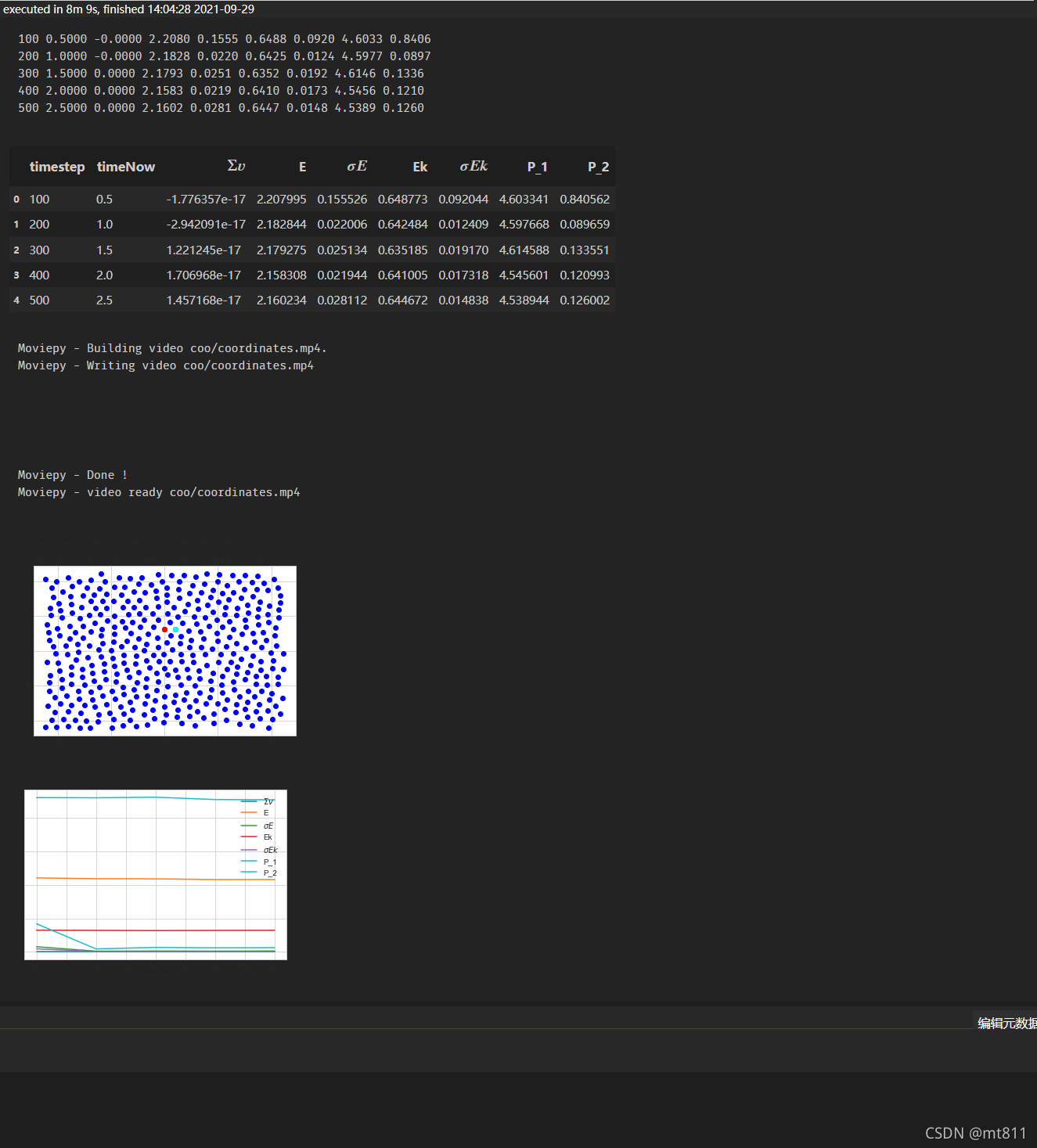
4. reference
- ����ģ��������������
-
Daan Frenkel ��Understanding Molecular Simulation:From Algorithms to Applications Second Edition��
-
��� �����Ӽ�����á�������ͼ��,���㷽����ģ�����ܡ�
-
����ģ����ʵ��
-
D.C RAPAPORT ��The Art of Molecular Dynamics Simulation 2th�� ���ij�����Ҫ�ο�����ڶ��½�
-
-
python��������ݷ���
-
��Python���:�����ŵ�ʵ����(�ڶ���)
-
��Python���ݿ�ѧ�ֲᡷ
5. ���
���ڳ�ѧ����˵�����Ƿ���ģ������Ļ������ۺͼ��㷽��,���DZ��ʵ��,����Ҫ���Ǵ�ȫ������,����ֱ��,����ϸ�ڿ���ʵ����ȥѧϰ,��Ҫ��ȥ���롣��˱��ĶԷ���ģ�����֪ʶ��ֻ�����н���,����ϵͳ��ϸ��֪ʶ��ο����������г����鼮�����ס���ע���ںš���������㻯ѧ��ѧ�ߡ� �ظ��ؼ���"MD_LJP" ���ɻ����زο�����,ģ������Լ�����ʾ���ļ���