NGS数据分析实践:06. 数据预处理 - 序列比对+PCR重复标记+Indel区域重比对+碱基质量重校正

一般变异识别之前需要进行数据预处理,包括序列比对、排序、PCR重复标记、Indel区域重比对和碱基质量重校正等步骤。以下详细介绍数据预处理的相关流程。
1. 序列比对
NGS测出来的短序列片段(read)存储于FASTQ文件里面(详见:NGS数据分析实践:03. 涉及的常用数据格式[1] - fasta和fastq格式)。Reads原本是来自于有序的基因组,但DNA经随机打断、建库和测序后,已经丢失了原来的有序性。因此,FASTQ文件中相邻的两条reads之间没有任何位置关系,它们都是来自于原本基因组中某个位置的随机短序列而已。
为了识别所测样本的基因组序列变异情况,需要先将FASTQ文件中的大量无序的reads,一条一条的去跟该物种的参考基因组作比较,找到每一条read在参考基因组上的位置,然后按顺序排列好(参考基因组的下载见:NGS数据分析实践:02. 参考基因组及注释库的下载)。这个过程被称为测序数据的序列比对,序列比对本质上是一个寻找最大公共子字符串的过程。常用的软件工具是bwa,它将BW(Burrows-Wheeler)压缩算法和后缀树相结合,能够以较小的时间和空间代价,获得准确的序列比对结果。
1.1 参考基因组建索引
首先,需要为参考基因组构建索引,以便能够在序列比对的时候进行快速的搜索和定位。
#下载hg38
nohup wget http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz &
gunzip hg38.fa.gz
# 1.1 对参考基因组用bwa构建索引
nohup time bwa index -a bwtsw -p hg38 hg38.fa 1>hg38.bwa_index.log 2>&1 &
# -p: 后面的hg38为索引文件的前缀名
# -a bwtsw: 参考基因组的大小大于2G时,需要加上该参数
cat hg38.bwa_index.log
## [bwa_index] 2205.20 seconds elapse.
## [bwa_index] Update BWT... 16.47 sec
## [bwa_index] Pack forward-only FASTA... 12.02 sec
## [bwa_index] Construct SA from BWT and Occ... 848.37 sec
## [main] Version: 0.7.17-r1188
## [main] CMD: bwa index -a bwtsw -p hg38 hg38.fa
## [main] Real time: 3200.623 sec; CPU: 3114.403 sec
1.2 序列比对
双末端测序(Pair-End Sequencing,简称PE测序)中,每一对的read1和read2都来自于同一个DNA片段,read1和read2之间的距离是这个DNA片段的长度,且read1和read2的方向刚好是相反的。read1在R1.fq文件中位置和read2在R2.fq文件中的位置是相同的,而且read ID之间只在末尾有一个’1’或者’2’的差别。

less -SN 20180629001_S1_L007_R1_001.fastq_clean.fq.gz
less -SN 20180629001_S1_L007_R2_001.fastq_clean.fq.gz
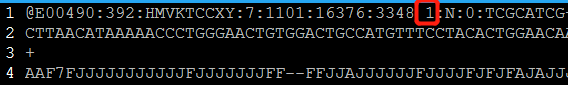
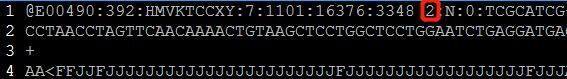
用法如下:
Usage: bwa mem [options] <idxbase> <R1.fq> [R2.fq]
# options说明:
# -t: 线程数
# -R:Read Group的字符串信息,非常重要,以@RG开头,用来将比对的read进行分组。对后续比对数据进行错误率分析、Mark duplicate和合并样本变异数据等步骤非常重要。
在Read Group中,有如下几个信息非常重要:
1) ID:Read Group的分组ID,一般设置为测序的lane ID(不同lane之间的测序过程认为是独立的),一般都包含在fastq文件名中。有时由于有些样本测得非常深,其测序结果需要经过多次测序(或者分布在多个不同的测序lane中)才能全部获得。
2) PL:所用的测序平台。在GATK中,PL只允许被设置为:ILLUMINA, SLX, SOLEXA, SOLID, 454, LS454, COMPLETE, PACBIO, IONTORRENT, CAPILLARY, HELICOS或UNKNOWN这几个信息。如果实在不知道,那么必须设置为UNKNOWN,名字不区分大小写。
3) SM:样本ID,有时候我们测序的数据比较多,可能会分成多个不同的lane分别测出来,这个时候SM名字就是可以用于区分这些样本;
4) LB:测序文库的名字,主要是为了协助区分不同的group而存在。文库名字一般可以在下机的fq文件名中找到,如果上面的lane ID足够用于区分的话,也可以不用设置LB。
除了以上这四个之外,还可以自定义添加其他的信息。这些信息设置好之后,在RG字符串中要用制表符(\t)将它们分开。
接下来,将reads比对至参考基因组,用小数据测试,无误再批量提交。
# 1.2 bwa比对
# Align the paired reads to reference genome hg38 using bwa mem.
# read长度≥70bp,推荐使用 bwa mem
bwa_idx=/your/index/path
cleandata_dir=/your/fq/path
ls $cleandata_dir
## 20180629001_S1_L007_R1_001.fastq_clean.fq.gz
## 20180629001_S1_L007_R2_001.fastq_clean.fq.gz
## 20180629002_S1_L007_R1_001.fastq_clean.fq.gz
## 20180629002_S1_L007_R2_001.fastq_clean.fq.gz
# 1) 单样本的序列比对
# 注:
# 参考基因组(含索引)使用前缀,不加扩展名。
# 否则报错[E::bwa_idx_load_from_disk] fail to locate the index files
# -R:设置短序列片段的标题行,该参数很重要,涉及到后续样本的合并等。
# 如报错[E::bwa_set_rg] the read group line is not started with @RG,需仔细核对该参数。
bwa mem -t 10 -k 32 -M $bwa_idx/hg38 \
-R "@RG\tID:20180629001\tLB:Targeted\tPL:Illumina\tSM:20180629001" \
$cleandata_dir/20180629001_S1_L007_R1_001.fastq_clean.fq.gz \
$cleandata_dir/20180629001_S1_L007_R2_001.fastq_clean.fq.gz \
1>$cleandata_dir/20180629001.sam \
2>$cleandata_dir/20180629001.bwa.align.log &
# 2) 多样本的批量序列比对
ls $cleandata_dir/*.fq.gz | awk -F'_' '{print $1}' | sort | uniq | while read id
do
#file=$(basename $id) #删除左边:路径
#pop=${file%%.*} #删除右边:点号及其后字符
#echo $pop;
nohup time bwa mem -t 10 -k 32 -M \
-R "@RG\tID:${id}\tSM:${id}\tLB:Targeted\tPL:Illumina" \
$bwa_idx/hg38/hg38 \
${id}_S1_L007_R1_001.fastq_clean.fq.gz \
${id}_S1_L007_R2_001.fastq_clean.fq.gz \
1>${id}.sam \
2>${id}.bwa.align.log &
done
2. 排序
注意,BWA比对后输出的SAM文件是没顺序的,排序步骤可使用samtools软件实现。SAM文件是文本文件,为了有效节省磁盘空间,一般会将它转化为二进制格式的BAM文件。排序和bam文件转换,可以同时进行。
# 2. sam to bam & sort
cd $cleandata_dir
for i in *.sam
do
file=$(basename $i)
id=${file%%.*}
echo $id;
nohup samtools sort -@ 5 -o ${id}.sorted.bam ${id}.sam 2>${id}.sorted.log &
done
# -@: 用于设定排序时的线程数
# -o:输出文件的名字
# -m 4G:限制排序时最大的内存消耗为4G
3. PCR重复标记
在NGS测序之前都需要先构建测序文库:通过物理(超声)打断或者化学试剂(酶切)切断原始的DNA序列,然后选择特定长度范围的序列去进行PCR扩增并上机测序。PCR扩增原本的目的是为了增大微弱DNA序列片段的密度,但由于整个反应都在一个试管中进行,因此其他一些密度并不低的DNA片段也会被同步放大,那么这时在取样去上机测序的时候,这些DNA片段就很可能会被重复取到相同的几条去进行测序。这可能会增大变异检测结果的假阴和假阳性率,详细说明见大神的文章:从零开始完整学习全基因组测序数据分析:第4节 构建WGS主流程。
因此,必须要进行PCR重复标记(去除)。Samtools的rmdup是直接将这些重复序列从比对BAM文件中删除掉;而Picard的MarkDuplicates默认情况则只是在BAM的FLAG信息中标记出来,而不是删除,这些重复序列依然会被留在文件中,可以在变异检测的时候识别到它们,并进行忽略。建议对PCR重复进行标记,留存这些序列,以便在需要的时候还可以对其做分析。
# index
# index文件的后缀名为.bai
bam_dir=/your/bam/path
ls $bam_dir/*.sorted.bam | while read i
do
file=$(basename $i)
id=${file%%.*}
# echo $id;
nohup samtools index $bam_dir/${id}.sorted.bam &
done
# 3. MarkDuplicates
# 把参数REMOVE_DUPLICATES设置为ture,那么重复序列就被删除掉,不会在结果文件中留存。
PICARD=/yourPath/picard-2.18.29-0
ls $bam_dir/*.sorted.bam | while read i
do
file=$(basename $i)
id=${file%%.*}
nohup java -jar $PICARD/picard.jar MarkDuplicates CREATE_INDEX=True \
# REMOVE_DUPLICATES=true \
I=$bam_dir/${id}.sorted.bam \
O=$bam_dir/${id}.sorted.markdup.bam \
M=$bam_dir/${id}.markdup_metrics.txt &
done
# index
# 为了可以随机访问这个文件中的任意位置,进而在后面进行“局部重比对”,需建立索引
bam_dir=/your/bam/path
ls $bam_dir/*.sorted.bam | while read i
do
file=$(basename $i)
id=${file%%.*}
# echo $id;
nohup samtools index $bam_dir/${id}.sorted.markdup.bam &
done
4. Indel局部区域重比对
Indel局部区域重比对的原因:①BWA和bowtie等全局搜索最优匹配的算法在存在Indel的区域及其附近的比对情况往往不是很准确,特别是当一些存在长Indel、重复性序列的区域或存在长串单一碱基(如,长串的TTTT或AAAAA等)的区域;②在这些比对算法中,对碱基错配和gap的容忍度是不同的。在read比对时,如果发现碱基错配和gap都可以的话,会更偏向于错配。这种偏向错配的方式,有时候还会反过来引起错误的gap。这就会导致基因组上原本应该是一个长度比较大的Indel的地方,被错误地切割成多个错配和短indel的混合集,这必然会让我们检测到很多错误的变异。且这种情况会随着所比对的read长度的增长而变得越加严重。
Indel局部区域重比对的目的:将BWA比对过程中所发现的有潜在序列插入或者序列删除(insertion和deletion,简称Indel)的区域进行重新校正,这个过程往往还会把一些已知的Indel区域一并作为重比对的区域。鉴于上述原因,重比对会减低后续变异识别的错误率。
此处需要用到GATK (GenomeAnalysisTK.jar)来实现,包含两个小步骤:①RealignerTargetCreator ,目的是定位出所有需要进行序列重比对的目标区域;②IndelRealigner,对所有在第一步中找到的目标区域运用算法进行序列重比对,最后得到新的结果。这两个步骤缺一不可,依次进行。需要指出的是,这里的-R参数输入的是完整文件名hg38.fa,而不是BWA比对中的索引文件前缀。
# 4. Indel局部区域重比对
# (1) RealignerTargetCreator,目的是定位出所有需要进行序列重比对的目标区域;
bam_dir=/your/bam/path
bwa_idx=/your/index/path
GATK=/yourPath/gatk-3.8/GenomeAnalysisTK.jar
DBSNP=/yourPath/annotation/dbSNP138/dbsnp_138.hg38.vcf.gz
Mills_indels=/yourPath/annotation/Mills1000G/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
KG_SNP=/yourPath/annotation/1000genomes/1000G_phase1.snps.high_confidence.hg38.vcf.gz
ls $bam_dir/*.sorted.markdup.bam | while read i
do
file=$(basename $i)
# echo $file
id=${file%%.*}
# echo $id;
nohup java -jar $GATK \
-T RealignerTargetCreator \
-R $bwa_idx/hg38.fa \
-I $bam_dir/${id}.sorted.markdup.bam \
-known $Mills_indels \
-o $bam_dir/${id}.IndelRealigner.intervals &
done
# 参考基因组需要.dict索引文件:samtools dict hg38.fa > hg38.dict
# (2) IndelRealigner,对所有在第一步中找到的目标区域运用算法进行序列重比对,最后得到新结果。
ls $bam_dir/*.sorted.markdup.bam | while read i
do
file=$(basename $i)
# echo $file
id=${file%%.*}
# echo $id;
nohup java -jar $GATK \
-T IndelRealigner \
-R $bwa_idx/hg38.fa \
-I $bam_dir/${id}.sorted.markdup.bam \
-known $Mills_indels \
--targetIntervals $bam_dir/${id}.IndelRealigner.intervals \
-o $bam_dir/${id}.sorted.markdup.realign.bam &
done
ll *.sorted.markdup.realign.bam | wc -l
5. 碱基质量重校正 (BQSR)
变异检测是一个依赖测序碱基质量值的步骤。碱基质量值是衡量我们测序出来的这个碱基到底有多正确的重要指标。它来自于测序图像数据的base calling,是由测序仪和测序系统来决定的。但影响这个值准确性的系统性因素有很多,包括物理和化学等对测序反应的影响、仪器本身和周围环境的影响等。因此,下机数据中的碱基质量值可能与真实值存在偏差。
碱基质量重校正 (Base Quality Score Recalibration, BQSR) 主要是通过机器学习的方法构建测序碱基的错误率模型,然后对这些碱基的质量值进行相应的调整。详细的解释依然见,从零开始完整学习全基因组测序数据分析:第4节 构建WGS主流程。
此处依然用GATK (GenomeAnalysisTK.jar)来实现,包含两个小步骤:①BaseRecalibrator,这里计算出了所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(sample.recal_data.table);②PrintReads,这一步利用第一步得到的校准表文件(sample.recal_data.table)重新调整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出新的BAM文件。
# 5. 碱基质量重校正 (BQSR)
#(1)BaseRecalibrator,计算所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(sample_name.recal_data.table)
ls $bam_dir/*.sorted.markdup.realign.bam | while read i
do
file=$(basename $i)
# echo $file
id=${file%%.*}
# echo $id;
nohup java -jar $GATK \
-T BaseRecalibrator \
-R $bwa_idx/hg38.fa \
-I $bam_dir/${id}.sorted.markdup.realign.bam \
--knownSites $Mills_indels \
--knownSites $DBSNP \
-o $bam_dir/${id}.recal_data.table &
done
ll *recal_data*
# (2) PrintReads,利用第一步得到的校准表文件(sample_name.recal_data.table)重新调整原来BAM文件中的碱基质量值,输出一份新的BAM文件。
ls $bam_dir/*.sorted.markdup.realign.bam | while read i
do
file=$(basename $i)
# echo $file
id=${file%%.*}
# echo $id;
nohup java -jar $GATK \
-T PrintReads \
-R $bwa_idx/hg38.fa \
-I $bam_dir/${id}.sorted.markdup.realign.bam \
--BQSR $bam_dir/${id}.recal_data.table \
-o $bam_dir/${id}.recal.bam &
done
ll *.recal.bam
注:因为BQSR实际上是为了(尽可能)校正测序过程中的系统性错误,因此,在执行的时候是按照不同的测序lane或者测序文库来进行的,这个时候@RG信息(BWA比对时所设置的)就显得很重要了,算法就是通过@RG中的ID来识别各个独立的测序过程的。
补充:有时由于有些样本测得非常深,其测序结果需要经过多次测序(或者分布在多个不同的测序lane中)才能全部获得。一般会先分别进行比对并去除(标记)重复序列和BQSR后再使用samtools进行合并,将同个样本的所有比对结果合并成唯一一个大的BAM文件。
Usage: samtools merge [-nurlf] [-h inh.sam] [-b <bamlist.fofn>] <out.bam> <in1.bam> [<in2.bam> ... <inN.bam>]
# -h FILE: Copy the header in FILE to <out.bam> [in1.bam]
# -b FILE: List of input BAM filenames, one per line [null]