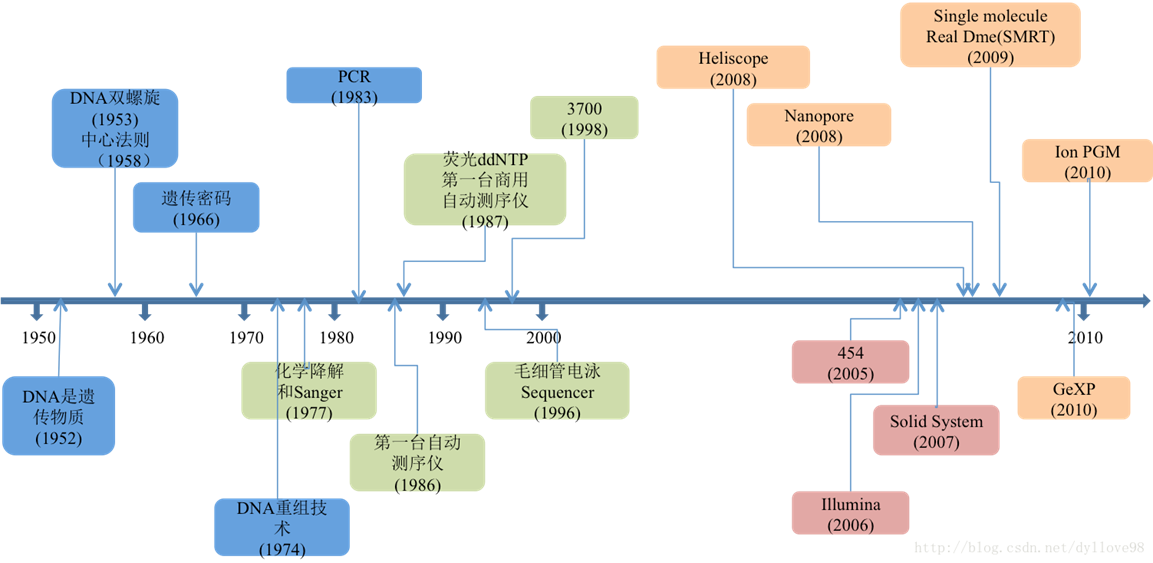
Ш§ДњВтађЪ§ОнЗжЮіЪЕеН
- жїНВШЫ:аьтљ
еуНДѓбЇвНбЇдКВЉЪП,ЕЯАВеяЖЯбаЗЂжааФЩњЮяаХЯЂЙЄГЬЪІ,ИКд№ NGS вХДЋМьВтЁЃ
БГОАНщЩм
Дг1977ФъЕквЛДњDNAВтађММЪѕ(SangerЗЈ)ЗЂеЙжСНёШ§ЪЎЖрФъЪБМф,ВтађММЪѕвбШЁЕУСЫЯрЕБДѓЕФЗЂеЙ,ДгЕквЛДњЕНЕкШ§ДњФЫжСЕкЫФДњ,ВтађЖСГЄДгГЄЕНЖЬ,дйДгЖЬЕНГЄЁЃЫфШЛОЭЕБЧАаЮЪЦПДРДЕкЖўДњЖЬЖСГЄВтађММЪѕдкШЋЧђВтађЪаГЁЩЯШдШЛеМгазХОјЖдЕФгХЪЦЮЛжУ,ЕЋЕкШ§КЭЕкЫФДњВтађММЪѕвВвбдкетвЛСНФъЕФЪБМфжаПьЫйЗЂеЙзХЁЃВтађММЪѕЕФУПвЛДЮБфИя,вВЖМЖдЛљвђзщбаОП,МВВЁвНСЦбаОП,вЉЮябаЗЂ,г§жжЕШСьгђВњЩњОоДѓЕФЭЦЖЏзїгУЁЃ
ЕкШ§ДњВтађММЪѕФПЧАвбОГЩЮЊПЦбаСьгђВЛПЩЛђШБЕФвЛжжжїСїММЪѕ,ЙуЗКгІгУгкЛљвђзщ DenovoЁЂШЋГЄзЊТМБОМьВтЁЂКъЛљвђзщЁЂжиВтађКЭБфвьМьВтЕШЖрИіЗНЯђ,ВЂЧвдкШОЩЋЬхНсЙЙБфвь(SV)ЕФМьВтжагазХВЛПЩЬцДњЕФгХЪЦЁЃ
ЕкШ§ДњВтађММЪѕФПЧАДцдкзХДэЮѓТЪНЯИпЕФЦПОБ,ЩњЮяаХЯЂбЇЗжЮіШэМўвВВЛЙЛЗсИЛ,ЕЋЪЧЮДРДЫцзХзМШЗЖШЕФЬсЩ§ЁЂЦНааВтађФмСІКЭУИЛюадЕШЮЪЬтЕФНтОі,ЕкШ§ДњВтађММЪѕЪЧЮДРДЗЂеЙЕФживЊММЪѕЧїЪЦ,ЪЕЯжДѓЙцФЃЩЬвЕЛЏНЋЪЧДѓЪЦЫљЧїЁЃ
ВтађММЪѕЗЂеЙРњГЬ
Ш§ДњВтађММЪѕНщЩм
ЕкШ§ДњВтађММЪѕЪЧжИЕЅЗжзгВтађММЪѕЁЃDNAВтађЪБ,ВЛашвЊОЙ§PCRРЉді,ВЛНіЪЕЯжСЫЖдУПвЛЬѕDNAЗжзгЕФЕЅЖРВтађ,ВЂЧвБмУтСЫЧБдкЕФPCRРЉдіДэЮѓКЭЦЋКУадЁЃ
ЕкШ§ДњВтађММЪѕФПЧАвбОГЩЮЊПЦбаСьгђВЛПЩЛђШБЕФвЛжжжїСїММЪѕ,ЙуЗКгІгУгкЛљвђзщDenovoЁЂШЋГЄзЊТМБОМьВтЁЂКъЛљвђзщЁЂжиВтађКЭБфвьМьВтЕШЖрИіЗНЯђ,ВЂЧвдкШОЩЋЬхНсЙЙБфвь(SV)ЕФМьВтжагазХВЛПЩЬцДњЕФгХЪЦЁЃ
Ш§ДњВтађММЪѕгХЕу
- ЖСГЄГЄ
- ВтађЫйЖШПь
- Ш§ДњВтађЩшБИаЁаЭЛЏ,БуНн,ЮШЖЈадКУ
Ш§ДњВтађММЪѕШБЕу
- ГЩБОЦЋИп
- ДэЮѓТЪНЯИп
- ЩњЮяаХЯЂбЇЗжЮіШэМўВЛЙЛЗсИЛ
ЧїЪЦ
ЮДРДЫцзХЕкШ§ДњВтађММЪѕЕФзМШЗЖШЬсЩ§ЁЂЦНааВтађФмСІКЭУИЛюадЕШЮЪЬтЕФНтОі,ЕкШ§ДњВтађММЪѕЪЧЮДРДЗЂеЙЕФживЊММЪѕЧїЪЦ,ЪЕЯжДѓЙцФЃЩЬвЕЛЏНЋЪЧДѓЪЦЫљЧїЁЃ
Ш§ДњВтађЦНЬЈМАдРэНщЩм
Ш§ДѓжїСїЦНЬЈ
| ВтађЗНЗЈ/ЦНЬЈ | ЙЋЫО | ЗНЗЈ/УИ | ЦНОљВтађГЄЖШ | ММЪѕдРэ |
|---|---|---|---|---|
| HeliScope | Helicos | БпКЯГЩБпВтађ/DNAОлКЯУИ | 30-35 bp | ЕЅЗжзггЋЙтВтађ |
| SMRT | Pacific Biosciences | БпКЯГЩБпВтађ/DNAОлКЯУИ | 100000 bp | ЕЅЗжзггЋЙтВтађ |
| Nanopore sequencing | Oxford Nanopore | ЕчаХКХВтађ/КЫЫсЭтЧаУИ | ЮоЯоГЄ | ФЩУзПзВтађ |
SMRT
PacBio SMRT(single molecule real time sequencing)ММЪѕгІгУСЫБпКЯГЩБпВтађЕФЫМЯы,ВЂвдSMRT аОЦЌЮЊВтађдиЬхЁЃ
ЛљБОдРэ:
DNA ОлКЯУИКЭФЃАхНсКЯ,4ЩЋгЋЙтБъМЧ4жжМюЛљ(МДdNTP),дкМюЛљХфЖдНзЖЮ,ВЛЭЌМюЛљЕФМгШы,ЛсЗЂГіВЛЭЌЙт,ИљОнЙтЕФВЈГЄгыЗхжЕПЩХаЖЯНјШыЕФМюЛљРраЭЁЃ
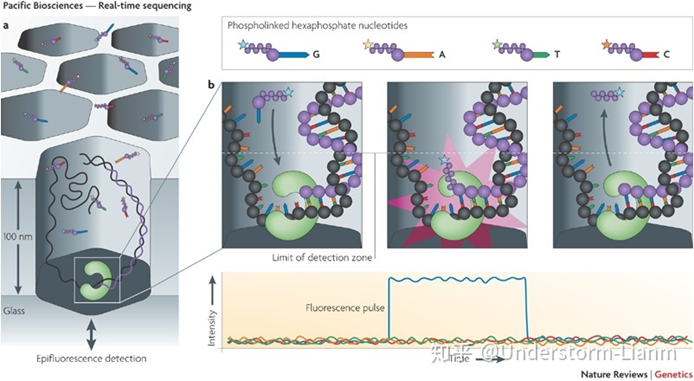
зЂ:DNA ОлКЯУИЪЧЪЕЯжГЌГЄЖСГЄЕФЙиМќжЎвЛ,ЖСГЄжївЊИњУИЕФЛюадБЃГжгаЙи,ЫќжївЊЪмМЄЙтЖдЦфдьГЩЕФЫ№ЩЫЫљгАЯьЁЃ
ВтађВНжш:
(1)ОлКЯУИВЖЛёЮФПтDNAађСа,УЊЖЈдкСуФЃВЈЕМПзЕзВП;
(2)4жжВЛЭЌгЋЙтБъМЧЕФdNTPЫцЛњНјШыСуФЃВЈЕМПзЕзВП;
(3)гЋЙтdNTPБЛМЄЙтееЩф,ЗЂГігЋЙт,МьВтгЋЙт;
(4)гЋЙтdNTPгыDNAФЃАхЕФМюЛљЦЅХф,дкУИЕФзїгУЯТКЯГЩвЛИіМюЛљ;
(5)ЭГМЦгЋЙтаХКХДцдкЪБМфГЄЖЬ,ЧјЗжЦЅХфМюЛљгыгЮРыМюЛљ,ЛёЕУDNAађСа;
(6)УИЗДгІЙ§ГЬжа,вЛЗНУцЪЙСДбгЩь,СэвЛЗНУцЪЙdNTPЩЯЕФгЋЙтЛљЭХЭбТф;
(7)ОлКЯЗДгІГжајНјаа,ВтађЭЌЪБГжајНјааЁЃ
гХЕу:
- ПЩвдЭЈЙ§МьВтЯрСкСНИіМюЛљжЎМфЕФВтађЪБМф,РДМьВтвЛаЉМюЛљаоЪЮЧщПі:
ШєМюЛљДцдкаоЪЮ,дђЭЈЙ§ОлКЯУИЪБЕФЫйЖШЛсМѕТ§,ЯрСкСНЗхжЎМфЕФОрРыдіДѓ,ПЩвдЭЈЙ§етИіРДжБНгМьВтМзЛљЛЏЕШаХЯЂЁЃ
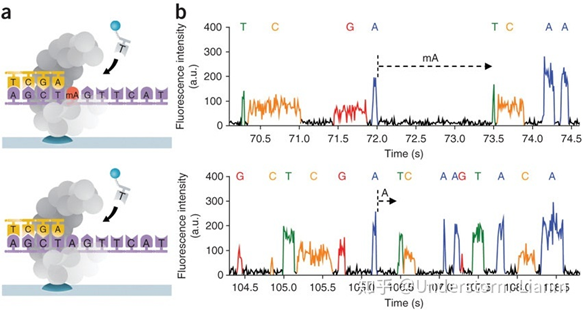
- ВтађЫйЖШКмПь,УПУыдМ10 ИіdNTP
- ЖСГЄГЄ
- ЮоашPCRРЉді,вВБмУтСЫгЩДЫДјРДЕФbias
- ашвЊЕФбљЦЗСПКмЩй,бљЦЗжЦБИЪБМфЛЈЗбЩй
ШБЕу:
- ВтађДэЮѓТЪБШНЯИп,ДяЕН15%,етМИКѕЪЧФПЧАЕЅЗжзгВтађММЪѕЕФЭЈВЁЁЃ?ГіДэЪЧЫцЛњЕФ,вђЖјПЩвдЭЈЙ§ЖрДЮВтађРДНјаагааЇЕФОРДэ
Nanopore sequencing
ЛљБОдРэ:
ФЩУзПзВтађЩшМЦСЫвЛжжЬиЪтЕФФЩУзПз,ПзФкЙВМлНсКЯгаЗжзгНгЭЗЁЃЕБDNA МюЛљЭЈЙ§ФЩУзПзЪБ,ЫќУЧЪЙЕчКЩЗЂЩњБфЛЏ,ДгЖјЖЬднЕигАЯьСїЙ§ФЩУзПзЕФЕчСїЧПЖШ(УПжжМюЛљЫљгАЯьЕФЕчСїБфЛЏЗљЖШЪЧВЛЭЌЕФ),СщУєЕФЕчзгЩшБИМьВтЕНетаЉБфЛЏДгЖјМјЖЈЫљЭЈЙ§ЕФМюЛљЁЃ
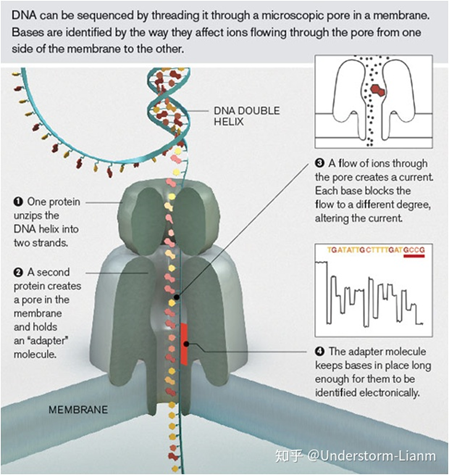
ВтађВНжш:
(1)НтТна§,НЋЫЋСДDNAНтПЊГЩЕЅСД;
(2)DNAЕЅСДЗжзгЭЈЙ§вЛИіПзЕРЕААз,ПзЕРжагаИіГфЕБзЊЛЛЦїЕФЕААзЗжзг;
(3)DNAЕЅЗжзгЭЃСєдкПзЕРжа,гавЛаЉРызгЭЈЙ§ДјРДЕчСїБфЛЏ,ЖјВЛЭЌЕФМюЛљДјРДЕФЕчСїБфЛЏЪЧВЛЭЌЕФ;
(4)зЊЛЏЦїЕААзЗжзгИаЪмВЛЭЌМюЛљЕФЕчСїБфЛЏ;
(5)ИљОнЕчСїБфЛЏЕФЦЕЦз,гІгУФЃЪНЪЖБ№ЫуЗЈЕУЕНМюЛљађСаЁЃ
ВтађЖСГЄ:
гЩгкВтађЮоашDNAОлКЯУИЕФСДЪНЗДгІ,ЫљвдВЛДцдкDNAОлКЯУИЕФЪЇЛюЮЪЬт,РэТлЩЯжЛвЊDNAЗжзгВЛЖЯПЊ,ОЭвЛжБПЩвдЭЈЙ§ФЩУзПз,ФПЧАдкЖдгкШЫКЭДѓГІИЫОњЕФВтађжаЙлВтЕНЕФreadЪЧ1MbзѓгвЁЃ
ВтађзМШЗТЪ:
NanoporeВтађзМШЗТЪКЭPacbioГжЦН,ЮЊ86%зѓгвЁЃЖјЧвЦ№ЪМЮЛжУе§ШЗТЪЦЋЕЭ,дкДѓдМ100ntЮЛжУДяЕНЮШЖЈ,ЧвДэЮѓЮЊЫцЛњВтађДэЮѓЁЃ
==ГіДэЪЧЫцЛњЕФ,вђЖјПЩвдЭЈЙ§ЖрДЮВтађРДНјаагааЇЕФОРДэ==
Ш§ДњВтађЪ§ОнЗжЮіСїГЬ
зЂ:вдЯТСїГЬвдМйЮЂаЭКЃСДдх(Thalassiosira pseudonana) Лљгк Nanopore sequencing ЕФЛљвђзщЪ§ОнЮЊР§ЁЃ
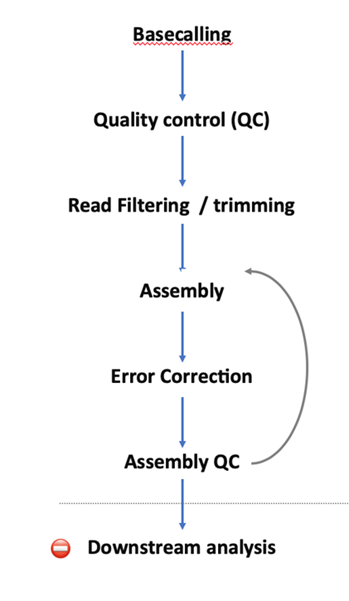
1. basecalling
- ФЩУзПзВтађвЧ MinION ЯТЛњЪ§ОнЮЊ fast5 ЖўНјжЦИёЪНЁЃ
- fast5ИёЪНДцДЂСЫnanoporeВтађЙ§ГЬжаШЋВПЕФЪфГіаХЯЂЁЃРяУцМЧТМзХЩшБИдЫааЪБШЋВПЕФаХЯЂ,АќРЈВЖЛёЕФЕчаХКХжЕ,ЩшБИдЫааЪБМф,ЕчбЙ,ЮТЖШЕШЕШаХЯЂЁЃ
MinION УПИібљБОЯТЛњЪ§ОнЪЧвЛЯЕСаЕФfast5ЮФМў,ШчЯТ:
Guppy
Guppy ЪЧЕБЧАЕФЁАЙйЗНЁБ ONT basecaller,ЛљгкbasecallingЕФЩёОЭјТчФЃаЭ,НЋдЪМЕчзгаХКХзЊЛЛГЩМюЛљ,ЩњГЩfastqИёЪНЁЃГ§СЫbasecallingжЎЭт,ЫќЛЙФмНјааЕЭжЪСПЕФreads Й§ТЫЁЂЛљгкХЃНђФЩУзПзВтађЕФadapterМєЧаЕШЙІФмЁЃ
ЪЙгУУќСюЪОР§:
guppy_basecaller
ЈCi /long-read-analysis/example_practice/0_rawdata
ЈCc /long-read-analysis/software/ont-guppy-cpu/data/dna_r9.4.1_450bps_hac
ЈCs /long-read-analysis/example_practice/1_basecalling
--num_callers 4
--cpu_threads_per_caller 3
ВЮЪ§ЫЕУї:
ЈCi АќКЌЫљгаfast5ЮФМўЕФФПТМ
-c ИљОнВтађаОЦЌКЭЪдМСКаЕФаЭКХбЁдёВЛЭЌЕФХфжУЮФМў,ХфжУЮФМўЮЛгкguppyЕФАВзАФПТМ/dataЯТ
ЈCs НсЙћЪфГіФПТМ
--num_callers basecalling БШНЯКФЪБ,ПЩНЋШЮЮёЗжВ№ЭЌЪБНјаа
--cpu_threads_per_caller УПИіШЮЮёЗжХфЕФ CPU ЯпГЬЩњГЩНсЙћШчЯТ:
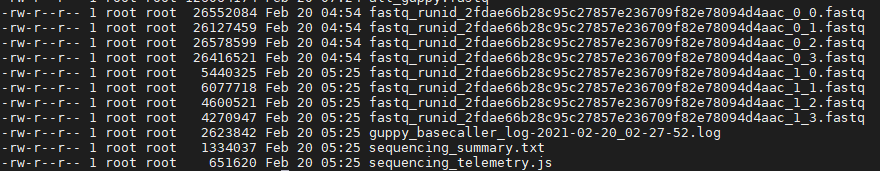
ШЛКѓНЋЫљгаfastqИёЪНЮФМўКЯВЂЕНвЛЦ№:
cat *.fastq > all_guppy.fastq2. Quality Control of Raw Reads
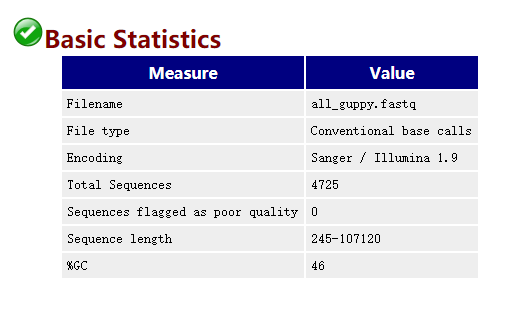
2.1 FastQC
FastQC ЪЧВтађЪ§ОнжЪПизюГЃгУЕФЙЄОпжЎвЛ,жЇГжIlluminaЁЂOxford NanoporeКЭPacBio data ЕШИїжжЦНЬЈЁЃ
ЪЙгУУќСюЪОР§:
fastqc
-o ./1_fastqc
/long-read-analysis/example_practice/1_basecalling/all_guppy.fastq
-t 4ДђПЊЩњГЩЕФНсЙћЮФМўall_guppy_fastqc.html:
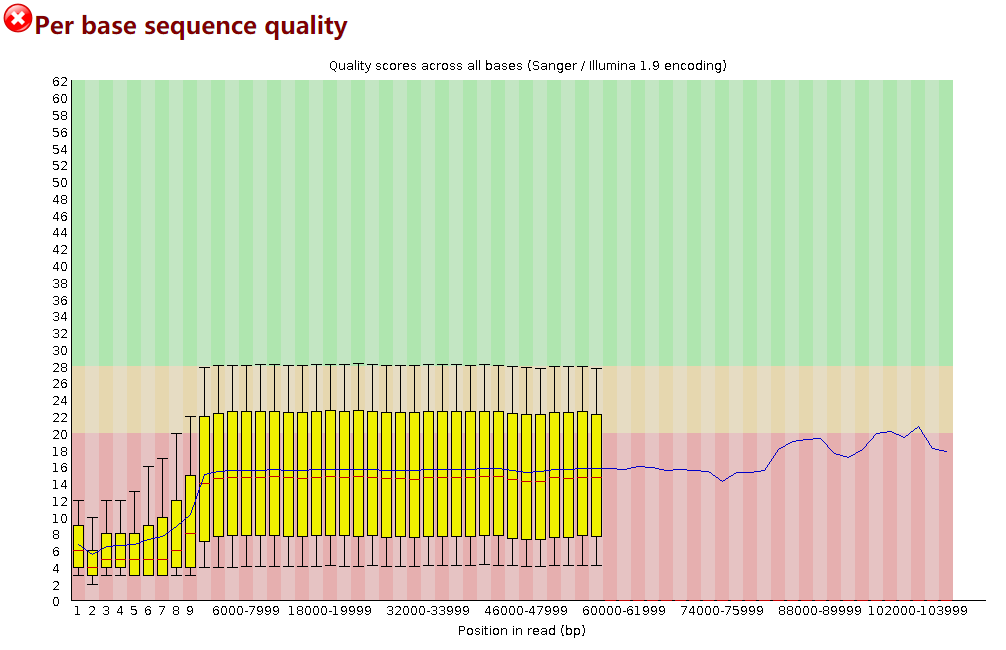
2.2 PycoQC
PycoQC ЪЧвЛжжЛљгкФЩУзПзЪ§ОнЕФЪ§ОнПЩЪгЛЏКЭжЪПиЙЄОпЁЃгыFastQCЯрБШ,ЫќашвЊвЛИіЬиЖЈЕФsequencing_summary.txt зїЮЊЪфШыЮФМў,ИУЮФМўгЩOxford nanopore basecaller (ШчGuppyЛђalbacore basecaller)ЩњГЩЁЃ
ЪЙгУУќСюЪОР§:
pycoQC ЈCf /long-read-analysis/example_practice/1_basecalling/sequencing_summary.txt ЈCo ./2_pycoQC/all_guppy_pycoQC.htmlДђПЊЩњГЩЕФНсЙћЮФМўall_guppy_pycoQC.html:
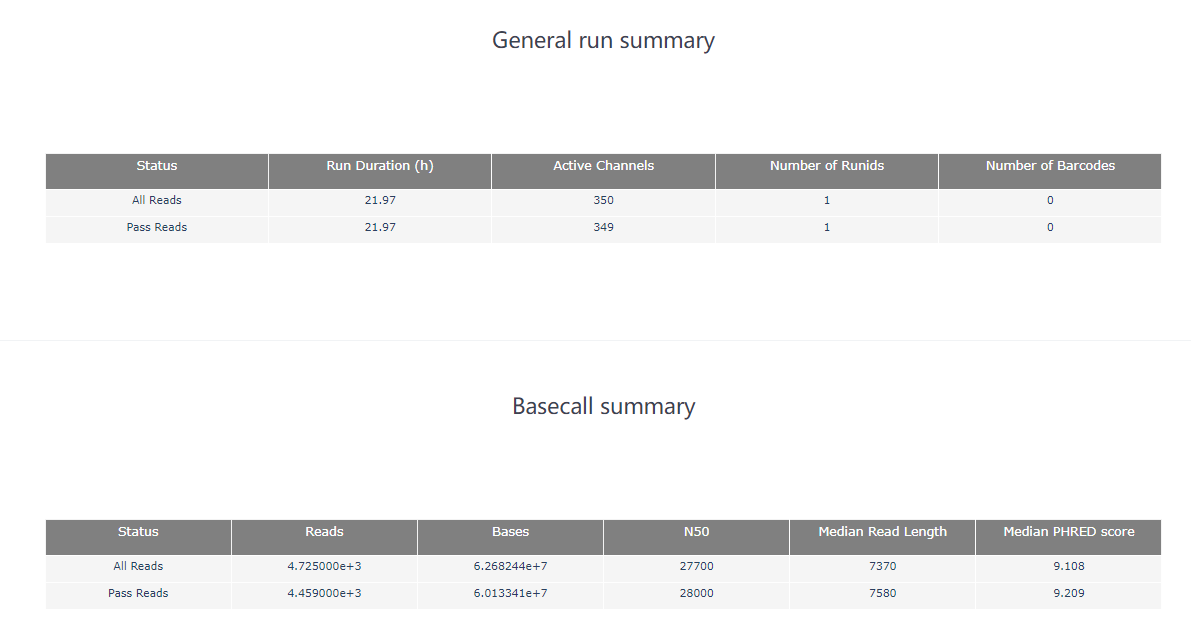
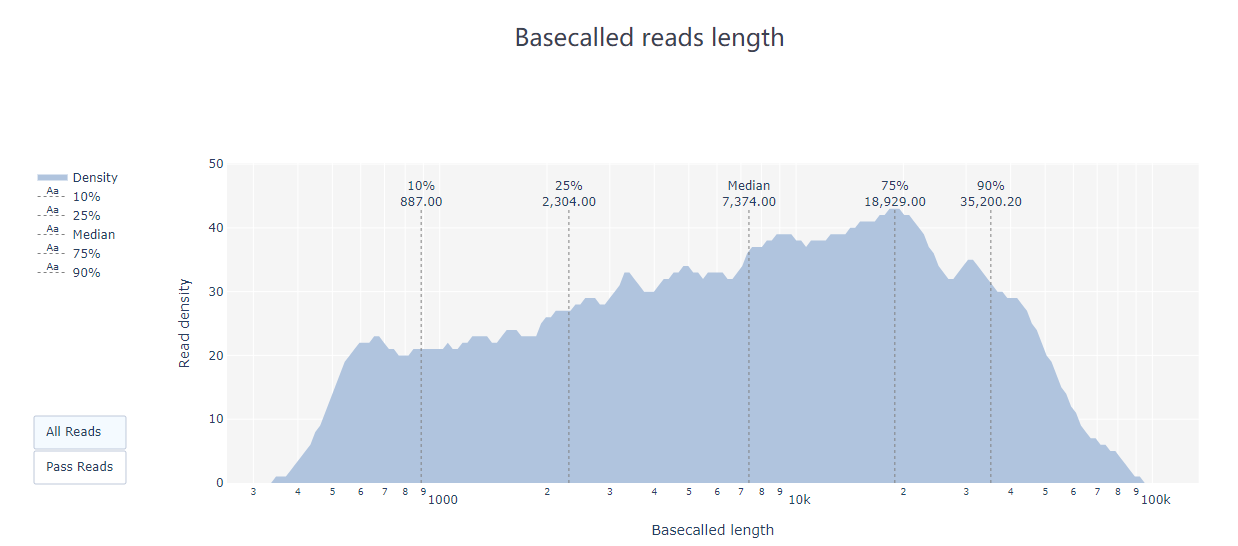
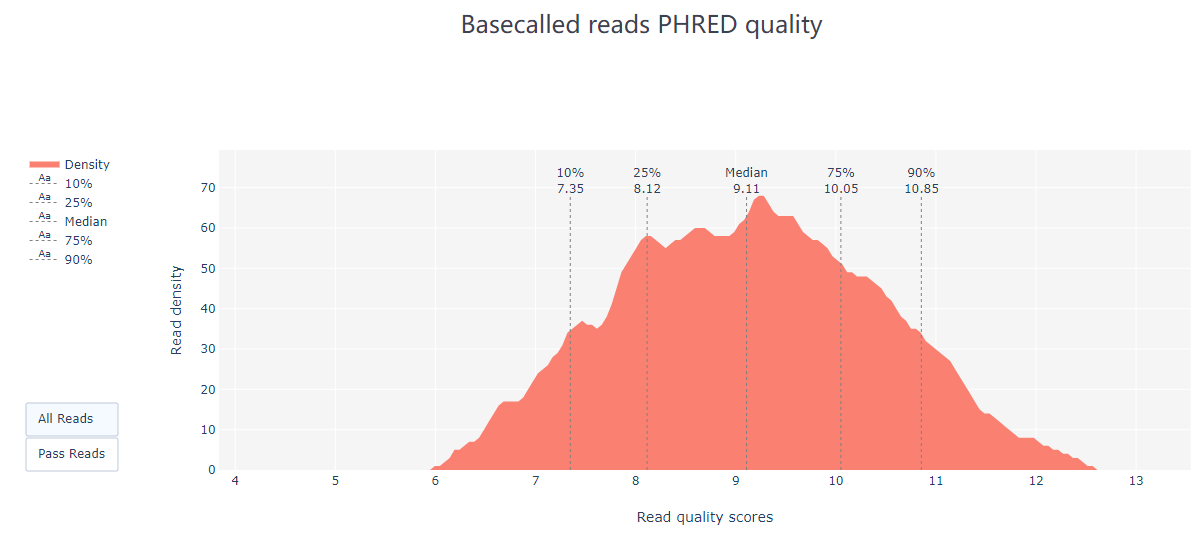
2.3 MinION_QC
MinIONQC вВЪЧвЛжжЛљгкФЩУзПзЪ§ОнЕФЪ§ОнПЩЪгЛЏКЭжЪПиЙЄОп,ашвЊвЛИіЬиЖЈЕФsequencing_summary.txt зїЮЊЪфШыЮФМўЁЃгыPycoQCЯрБШ,ЫќФмЙЛБШНЯЖрИіВтађНсЙћЕФжЪПиНсЙћЁЃ
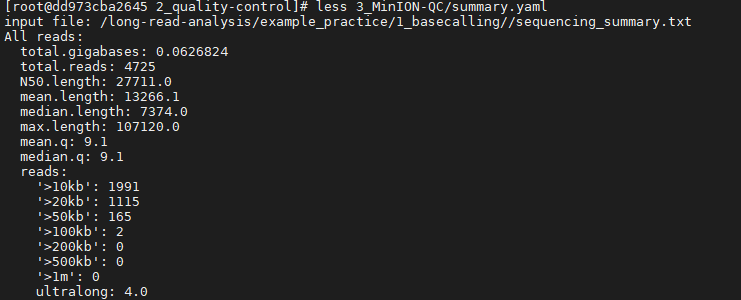
MinIONQC.R ЈCi /long-read-analysis/example_practice/1_basecalling/sequencing_summary.txt -o ./3_MinION-QCВщПДНсЙћЮФМўsummary.yaml:
3. Filtering, trimming and adapter removal
-
Adapter Removal using PoreChop
ЪЙгУУќСюЪОР§:
porechop
ЈCi /long-read-analysis/example_practice/1_basecalling/all_guppy.fastq
-o ./porechopped.fastq
--discard_middle-
Read trimming and filtering using NanoFilt
ЪЙгУУќСюЪОР§:
NanoFilt -l 500 --headcrop 10
./porechopped.fastq
> ./nanofilt_trimmed.fastq4. Genome Assembly
гЩгкЕкШ§ДњВтађММЪѕЕФИпЮѓВюТЪ,НЋPacBioКЭOxford NanoporeЕШГЄЖСГЄЪ§ОнзщзАГЩ contigs ЖдЦеЭЈЕФЕкЖўДњВтађзщзАШэМўРДЫЕЪЧвЛИіЬєеНЁЃдкЙ§ШЅЕФМИФъРя,дНРДдНЖрЕФзЈУХЮЊГЄЖСГЄreads ЩшМЦЕФзщзАШэМўБЛЗЂВМ,Р§ШчCanuЁЂFlyeЁЂShastaКЭminiasmЁЃ
ВЛЭЌЕФзщзАШэМўЖдВЛЭЌЕФЛљвђзщгаВЛЭЌЕФзїгУЁЃЛљвђзщДѓаЁЁЂжиИДадЁЂGCКЌСПЕШвђЫиЖМЛсгАЯьзщзАШэМўЕФадФмЁЃзюКУЕФАьЗЈЪЧдЫааЖрИізщзАШэМў,ШЛКѓБШНЯНсЙћ,ОЦРЙРКѓОіЖЈЪЙгУФФвЛИіШэМўЕФНсЙћЁЃ
вдЯТНЋНщЩм minimap2-miniasm ЕФЛљвђзщзщзАСїГЬ:
Genome Assembly with Minimap2 and Miniasm
minimap2-miniasm СїГЬЪЧзщзАГЄЖСГЄreadЕФвЛжжЗЧГЃПьЫйКЭИпаЇЕФЗНЗЈ,ИіШЫБШНЯЭЦМіЁЃ
ЪЙгУУќСюЪОР§:
#ЪЙгУ minimap2 ЖдађСаНјааздЮвБШЖд,ЩњГЩoverlapаЃе§ЮФМў
minimap2 ЈCx ava-ont
/long-read-analysis/example_practice/3_filtering-trimming/porechopped.fastq/nanofilt_trimmed.fastq
/long-read-analysis/example_practice/3_filtering-trimming/porechopped.fastq/nanofilt_trimmed.fastq
| gzip -1 > ./minimap.paf.gz
#ЪЙгУ miniasm НјааађСазщзА
miniasm -f
/long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq
./minimap.paf.gz > miniasm.gfa
#ЬсШЁcontigsађСаБЃДцЮЊfastaИёЪН
awk '/^S/{print ">"$2"n"$3}' miniasm.gfa > miniasm.fasta
5. Error correction
5.1 Error Correction using Racon
Racon ШэМўЪЧЮЊСЫВЙГфminimap2/miniasm СїГЬЖјПЊЗЂЕФ,ЕЋПЩвдгУгкШЮКЮГЄЖСГЄreadsЖСШЁЕФзщзАНсЙћЁЃЫќЬсЙЉСЫвЛИіПьЫйЕФвЛжТадЫуЗЈ,ПЩЖдЖўДњЖЬreads КЭ Ш§ДњГЄЖСГЄreads НјаааЃе§ЁЃ
ЪЙгУУќСюЪОР§:
minimap2
/long-read-analysis/example_practice/4_genome-assembly/miniasm.fasta
/long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq
> ./minimap.racon.paf
racon
/long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq
minimap.racon.paf
/long-read-analysis/example_practice/4_genome-assembly/miniasm.fasta
> ./miniasm.racon.consensus.fasta
5.2 Error Correction using Minipolish
гы Racon РрЫЦ,Minipolish ЪЧзЈУХЮЊаЃе§ minimap2/miniasmСїГЬЕФНсЙћЖјБраДЁЃЪТЪЕЩЯ,minipolish ЪЧЕїгУ Racon РДгХЛЏ miniasm ЕФНсЙћ,ЕЋгы Racon ВЛЭЌЕФЪЧ,ЫќЖСШЁКЭЪфГіЮФМўЪЧminiasm ЕФGFAИёЪН,ЖјВЛЪЧfasta ИёЪНЁЃ
ЪЙгУУќСюЪОР§:
minipolish -t 4
/long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq
/long-read-analysis/example_practice/4_genome-assembly/miniasm.gfa
> ./minipolished_assembly.gfa
awk '/^S/{print ">"$2"n"$3}' minipolished_assembly.gfa > minipolished_assembly.fasta
5.3 Pilon
PilonПЩвддкRacon жЎКѓдЫаа,ЭЈЙ§ОРе§ВхШы/ШБЪЇ(Indel)КЭЕЅКЫмеЫсЖрЬЌад(SNPs) ЕФДэЮѓНјвЛВНЬсИпзщзАжЪСПЁЃ
- Pilon
ЪЙгУУќСюЪОР§:
# index the consensus sequence
bwa index /long-read-analysis/example_practice/5_error-correction/1_Racon/miniasm.racon.consensus.fasta
# map reads
bwa mem -t 5
/long-read-analysis/example_practice/5_error-correction/1_Racon/miniasm.racon.consensus.fasta
-x ont2d
/long-read-analysis/example_practice/1_basecalling/all_guppy.fastq
> ./bwa_mapping.sam
samtools view -Sb bwa_mapping.sam > bwa_mapping.bam
samtools sort -o bwa_mapping.sorted.bam bwa_mapping.bam
samtools index bwa_mapping.sorted.bam
# run Pilon
java -Xmx16G -jar /long-read-analysis/software/pilon/pilon-1.23.jar
--genome /long-read-analysis/example_practice/5_error-correction/1_Racon/miniasm.racon.consensus.fasta
--bam bwa_mapping.sorted.bam
6. Variant calling
ГЄЖСГЄВтађЕФЫфШЛПЩвдМьВтЕЅКЫмеЫсЖрЬЌад(SNPs),ЕЋЪЧдкМьВтГЄЦЌЖЮНсЙЙБфвь(SVs)ЗНУцИќОпгХЪЦЁЃШ§ДњВтађММЪѕЕФНЯИпДэЮѓТЪ,ЪЙЕУЖдЕЅКЫмеЫсЖрЬЌадЕФМьВтОпгаКмИпЕФЬєеНадЁЃЕНФПЧАЮЊжЙ,жЛгаКмЩйЕФЙЄОпФмЙЛЖдЕкШ§ДњВтађЪ§ОнНјаа SNP КЭ SV ЕФМьВтЁЃ
Sniffles
Sniffles жївЊгУгкМьВтГЄЖСГЄЪ§ОнЕФSV,зЈУХЮЊ Pacbio КЭ Oxford NanoporeЪ§ОнЩшМЦ,вбОеЙЯжГіСЫСМКУЕФадФмЁЃ
ЪЙгУУќСюЪОР§:
# map to genome
minimap2 --MD -a /long-read-analysis/example_practice/Thalassiosira-pseudonana.chr17.fasta
/long-read-analysis/example_practice/1_basecalling/all_guppy.fastq
> mapped.sam
# Convert to bam file
samtools view -bS mapped.sam > mapped.bam
# Sort the bam file
samtools sort -o mapped.sorted.bam mapped.bam
# create an index file
samtools index mapped.sorted.bam
# Run sniffles
sniffles -m mapped.sorted.bam -v variants.vcfВщПДНсЙћЮФМўvariants.vcf:
ЕкШ§ДњВтађММЪѕеЙЭћ
ЕкШ§ДњВтађММЪѕФПЧАвбОГЩЮЊПЦбаСьгђВЛПЩЛђШБЕФвЛжжжїСїММЪѕ,ФПЧАвбОдкШОЩЋЬхНсЙЙБфвь(SV)ЕФМьВтжагазХВЛПЩЬцДњЕФгХЪЦЁЃ
ЫфШЛЕкШ§ДњВтађММЪѕФПЧАДцдкзХДэЮѓТЪНЯИпЕФЦПОБ,ЩњЮяаХЯЂбЇЗжЮіШэМўвВВЛЙЛЗсИЛ,ЕЋЪЧЮДРДЫцзХзМШЗЖШЕФЬсЩ§ЁЂЦНааВтађФмСІКЭУИЛюадЕШЮЪЬтЕФНтОі,ЕкШ§ДњВтађММЪѕЪЧЮДРДЗЂеЙЕФживЊММЪѕЧїЪЦ,ЪЕЯжДѓЙцФЃЩЬвЕЛЏНЋЪЧДѓЪЦЫљЧїЁЃ
Ш§ДњВтађЪ§ОнЗжЮізЈЬт
- ФкШн:Ш§ДњВтађЪ§ОнЗжЮіЪЕеН
- жїНВШЫ:аьтљ
- ЪБМф:2021Фъ3дТ06Ше ЯТЮч 2:00-5:00
- ЕиЕу:ЯпЩЯ?