�����š�ȫ�������������(GWAS)ԭ��
���µ�����/ͼƬ/���벿��/ȫ����Դ�����ѧ������,���»�������ɸ���,�������ѧϰʹ�á�
Ŀ¼
1.3 Hardy-Weinberg����&��������
1.ǰ��֪ʶ����
1.1 ��С���˷�


?
1.2 GWAS����ѧԭ��

?a�����Ż�Ϊ 2.8387, b�����Ż�Ϊ 2.0968 ,��ʽ y ?= ?2.8387* x ?+ 2.0968
ʵ����,�����ڼ����ʱ��,����������ı���,�����Ա�,����,Ʒϵ�ȡ���Щ����Ҳ��Ӱ����͵ı��������,������������������ʱ,���㹫ʽ�����ı�һ��:y ?= ?ax ?+ z�� ?+ ?b
y:�о��ı���
x:����������,����ָÿһ��SNP
a:SNP��ϵ��
z:����,�Ա������
��:����,�Ա�����ص�ϵ��
b:�в�,�������������SNP,�Ա���������������������Ӱ����͵�����;
1.3 Hardy-Weinberg����&��������

?
1.4 ������ƽ��
��Ⱥ���Ŵ�ѧ�о���,LD������ƽ������Ƿdz�����������,ͬʱҲ�ǹ��������Ļ��������������ֻҪ������������ȫ�����Ŵ�,�ͻ���ֳ�ij�̶ֳȵ�����,��������ͽ�������ƽ�⡣
�������ڵĻ���A B, ���Ǹ��Եĵ�λ����Ϊa b. ����A B������Ŵ�,����Ⱥ���й۲�õ��ĵ���������� AB �г��ֵ�P(AB)�ĸ���Ϊ P(A)*P(B)��
������ʵ�ʹ۲�õ�Ⱥ���е���������� AB ͬʱ���ֵĸ���ΪP(AB)���������Ե�λ�����Ƿ������ϵ�,��������λ��������ȫ������,��P(AB)=P(A)*P(B)��
�����Ƿ���ʵ����P(AB)��P(A)*P(B),��˵��A B��������ƽ��ġ�
��ƽ��̶ȶ�����ָ��ΪD:D=P(AB)- P(A) *P(B)
����λ��ͬһȾɫ���������λ����(AB)ͬʱ���ڵĸ��ʴ�����Ⱥ��������ֲ���ͬʱ���ֵĸ���,�������㴦��LD״̬��
1.5 ������ͼ
������ͼ(manhattan plot)��һ��ɢ��ͼ,ͨ��������ʾ���д������ݵ�,�����������������ֵ�ֲ������ݡ�
��ͼ��ÿ�������һ��SNP,����Ϊÿ��SNP���������P valueȡ-log10,����ΪSNP���ڵ�Ⱦɫ��,ͼ�жԺ�ѡλ��ķֲ�����ֵһĿ��Ȼ��
����λ���P valueԽС��-log10(P value)Խ��,��λ���������״���ȹ����̶�Խǿ��
����ͨ����˵�ܵ�������ƽ���Ӱ��,ǿ����λ����Χ��SNPҲ����ʾ����Խϸߵ��ź�ǿ��,�����������ߵݼ�,���Ի������ͼ�к�ɫ���ֵ�����
һ��,��GWAS���о���,P value����ֵ��10^-6 ����10^-8���¡�

ggplot��ע��SNP

ע��:������ͼ��,������SNP�����Ǻ�����Ⱥ��ð����,�����ƺ�����������,�����¥����һ��,�ӵ�����ð�����ġ���һ��������ʵ����������һ���SNP,���кܸߵ�LD score����Ȼ������ͼ��ÿ������SNP,����ͨ���������������SNPָ��ij������,��Ϊ������ע�Ļ���SNP���²���Դ,�������ҳ�����ֻ����������SNP��
ע��:��ͻ����SNP���п��ܲ��ǹؼ���SNP,��ֻ���ٽ��ؼ�SNP��λ�㡣
?
1.6 ��ʽͼBox-plot
����ͼ(Box-plot)��һ��������ʾһ�����ݷ�ɢ�����ͳ��ͼ,����״�����Ӷ���������Ҫ���ڷ�ӳԭʼ���ݷֲ�������,���ҿ��Խ��ж������ݷֲ������ıȽϡ�
����ͼ����ʾ��һ�����ݵ����ֵ(Maximum)����Сֵ(Minimum)����λ��(Median)�������ķ�λ��(1st/3rd Quartile),ͬʱ��������ʾ�ݳ�ֵ(Outlier)��
?
��һ�ķ�λ��(Q1),�ֳƽ�С�ķ�λ��,���ڸ�������������ֵ��С�������к��25%�����֡�
�ڶ��ķ�λ��,�ֳ���λ��,���ڸ�������������ֵ��С�������к��50%�����֡�
�����ķ�λ��(Q3)�ֳƽϴ��ķ�λ��,���ڸ�������������ֵ��С�������к��75%�����֡�
�ݳ�ֵ,�Ǹ����ķ�λ���(interquartile range)���м����:
�ķ�λ���= Q3-Q1=��Q,������ [Q3+1.5��Q, Q1-1.5��Q] ֮���ֵ������Ϊ�ݳ�ֵ��
1.7 QQ plot
Q-Q(��λ��-��λ��)ͼ,��ͨ���������������ʷֲ��ķ�λ�����Ƚ����߷ֲ���ϵ��ͼ�η�����Q-Q plot�����ڱȽ����ݼ��ϻ����۷ֲ���
- Q-Q plot��������ֵ��ʵ��ֵ�Ĺ�ϵͼ,x=����ֵ,y=ʵ��ֵ��
- ����״̬��y=x,��Щ��Ӧ������y=x����һ����0��,б��45�ȵ�ֱ���ϡ�
- ���ʵ�ʵ��������ֵ���ϵ���һ�����̬�ֲ�,mean=��,�����ĵ��Ӧ������y= x+������ֱ����,��Ϊ��ʱÿ��ʵ�ʵ��������ֵ,Ӧ�þ�����������ֵ���� x+�����������Ա仯�õ��ġ�
- ���������ȷʵȡ��һ����̬�ֲ�, ��ô����������Խ��,QQplot��Խ�ӽ�һ��ֱ��,��֮,����������������ʱ,QQplot�еĵ������ƫ��ֱ�ߡ�
?
?
2��GWAS�Ķ���
2.1 ������Ҫ֪���ĸ���:
������:��ָijһ�������ȫ��������ϵ��ܳơ�����ӳ��������Ŵ�����,����˫��õ�ȫ��������ܺ͡�GWAS����Ҫ��ע����ȫ�����鷶Χ�ϵ��Ŵ�����(SNP)��
SNP:SNP���������̬������common��genetic variation���͡�
���䲢�еĻ���С��indel(�������ɾ��һ��С����,һ����50bp����)��CNV(����������)��SV(��Ľṹ����,һ����50bp���ϵij����еIJ��롢ɾ����Ⱦɫ�嵹λ��)��
����:�������ɹ۲����״��
Allele frequency��λ����Ƶ��:ÿһ������λ�㶼����������allele,��ͬallele֮�������Ե�Ƶ���ϵIJ���,���������A��a�������ʵ�Ƶ��,�������Ǽ��λ��,��������״����
2.2?ȫ�������������
ȫ�������������(Genome wide association study,GWAS)�ǶԶ��������ȫ�����鷶Χ���Ŵ�����(���)��̬�Խ��м��,��û�����,��������������ɹ۲����״,������,����Ⱥ��ˮƽ��ͳ��ѧ����,����ͳ������������ p ֵɸѡ�����п���Ӱ�����״���Ŵ�����(���),�ھ�����״������صĻ���
�ܶ�ط����ǰ��������һ�λ������,�����֮������ȫ�����鷶Χ,�о���Ŀ����״�����ĺ�ѡ������������������֤�ͷ���,�ٽ�һ�����;��ǿ�ij��SNP��case��control������Ⱥ���Ƿ���allele frequency���������졣
�����������GWAS�Ĵ������̺Ͳ���:

?����Ҳ���˽�GWAS������ͼ��������:

?
������������Դ:GWASͨ����Ҫ����������,��ǧ�DZ���,���پ�̫��,�����еĶ��ﵽ�˼���������ʮ����GWAS��һ��������WGS(��̫����),������DNA chip��DNAоƬ(SNP array),ֻ�����˳�����10^6��SNP�����Ѷ�һЩ����WES,Ǯ�ܶ�Ļ�������WGS,�������ͷDZ�����������
����������Դ:��������һ���ǿ����о���ǰ��ʵ�鶯��Ⱥ��������õ�����,����־Ը����ļ������,����Ҳ�кܶ�������ݡ�
3��GWAS��������Ԥ����

?
3.1�ʿص�ԭ��:
һ����˵,һ���˵Ļ���������һ����������ı��,��˺��ٴ���ͬʱӰ�������Ӱ������͵ı��졣Ҫȥ��һЩ��������ƫ�������(Ҳ���������ڵ�һ�������ᵽ�IJв�),��Ҫ��Ⱥ��ṹ����������ѪԵ��ϵ�������Բ�����
?
3.2���������ݵ��ʿ�:
�ų�������ȱʧ�ʸߵĸ��塣
�ų������Ӻ���̫�ߺ�̫�͵����ݡ�

?����ͼ,����SNP������ֵ���趨,����²���-log(P value)��������ȱʧ��(call rate)>0.05����������MAFֵ<0.01��SNPλ������

?��ͼΪ�ܽ���ļ�������������Ҫ��ɵ�task,���,��ó��ɾ���SNP������:
| Step | Command | Function |
| Missingness of SNPs | �\�\geno | ���кܵ�genotype calls��SNP�����������������ж�ȱʧ��SNP���ų��ˡ� |
| Missingness of individuals | �\�\mind | ���кܵ�genotype calls�ĸ��屻������������ȱʧ�ʹ��ߵĸ������ݱ����� |
| Sex discrepancy | �\�\check�\sex | ���� X Ⱦɫ���Ӻ�/�����ʼ�����ݼ��м�¼�ĸ�����Ա������Ա�֮��IJ��졣 |
| Minor allele frequency (MAF) | �\�\maf | �������������õ� MAF(�ε�λ����Ƶ��) ��ֵ�� SNP�� |
| Hardy�CWeinberg equilibrium (HWE) | �\�\hwe | �ų�ƫ�� Hardy-Weinberg ƽ��ı�ǡ� |
| Heterozygosity | �ų������Ӻ���̫�ߺ�̫�͵����ݡ� | |
| Relatedness IBD | �\�\genome | Calculates identity by descent (IBD) of all sample pairs. �������������Ե�IBDֵ��IBD:ѪԴͬһ�� |
| Relatedness | �\�\min | ������ֵ����������Ը�����ѡ��ֵ�ĸ����б��� ����ζ�ſ��Լ����� pi-hat >0.2 ��ص�������(����������)�� |
| Population stratification IBD | �\�\genome | �������������Ե�IBDֵ��IBD:ѪԴͬһ�� |
| Population stratification | �\�\cluster �\�\mds�\plot?k | ���� IBS �����������κ��ӽṹ�� k ά��ʾ�� |
?
3.4���������ʿ�:
��Ҫ�Ա�������ͳ�Ʒ���:
��������Ϊ��Ԫ����ʹ�����ع�
��������Ϊ�����Ա�������ʹ�����Իع�
����������̬����(���������̬�ֲ�,��ת������Ϊ��̬�ֲ�)
�������ݾ�ֵ����ֵ�����ֵ����Сֵ,Ӱ�����ӶԱ��͵�Ӱ������ȵȡ�
?��Щ�ع�,��̬����,�����Сֵ,���Ǻܻ�������ѧ��������չ����
?
3.5��������ת
ʲô����������ת:�ںϲ����ݹ��̵���,�����ᷢ�ֲ�ͬ��Դ����������������ͳһ�ġ�
�Ӹ���ĽǶ�����:�����С����С��ֱ��õ���һ�����������ݡ���ô�������¼��ֿ���:1)С���Ļ���������ͳһ���������߸���;2)С��Ļ���������ͳһ���������߸���;3)С����С�춼��֪�����ǵ�������û��ͳһ��,���������ø�����,���Լ������
�ڲ�֪�������������������Ƿ�ͳһ�������,���ֱ�Ӻϲ���ͬ��Դ������:
���ڴ����ͻ��λ��,�ϲ���û�������,�������A/G,A/C,T/G,T/Cͻ���ǿ��Թ�ܵ�,������A/T,C/G����ͻ��õ��Ľ����:����ͬһ������,����С�������ݿ���ʾ�ļ����A,��С������ݿ���ʾ����T��
��Ⱥ��ĽǶ�����:����ͬһ������,����С�������ݿ���ʾ�ļ����A,��С������ݿ���ʾ����T��������������ǵĻ���
�������:snpflip��������ƪblog��ʱ���漰ʵ������,ʵ�ٲ�����漰��������������͵���������,�ᵥ����дһƪ��(�����漰����ʵ�ֵ�)
3.6 �������
����:�������(genotype imputation),���Ǵ���оƬ����ʱ(ǰ��������Դ���ֵ�����DNA chip�ķ�����õ�����),�����ߵ�һ�����̡��������WGSȫ�������������,��ô�Ͳ���Ҫimputation��
����������������:��ȷ��Ԥ��û�б�оƬ��������ǵ�SNPλ��Ļ�����,ʹ�ø�����Ŵ�λ��Ӧ�õ�����������,�Ӷ���߷����µ��²�����Ŀ����ԡ�
�ҵ�����:Ҳ����˵������ûǮ������IJ������ʱ,��ЩSNP�DZ���©��,���������ģ������������䡣���ģ�����ɸ��ܶ�SNP���ɵĵ�����(haplotypes)���ο�ģ�塣����ǧ�˻�����ƻ������,����7ǧ��Ķ�̬��λ�㱻����,�ɴ˹�����һ�ŷḻ�������Ŵ�������ͼ��,Ϊ����������ṩ���������ݡ�
����������ԭ����������:
- �ھ�����ͬ�����͵ļ�ϵ��,�Ŵ�����ٵ��������Բ����Ŵ���Ƕ���������л�������䡣
- ����û����Ե��ϵ������,��������Ҳ��������,��Ҫ�IJ��������ѪԵ��ϵ������֮�乲���ĵ����ͱȼ�ϵ����֮���Ҫ�̺ܶࡣ
- ������Ե��ϵ�������л����������Ҫһ�����ܶ��Ŵ���ǹ��ɵ�������ͼ����Ϊ���ա�ͨ���Աȴ���������Ͳο�ģ��,�ҵ�����֮�乲�еĵ�����,Ȼ��Ϳ��Խ�ƥ���ϵIJο�ģ���е�λ�㸴�Ƶ�Ŀ�����ݼ��С�
- Ȼ��,�������еĵ�������Զ�ȷһ�¡�����ȷ�϶���һ��������Ӧ�ñ����,ͨ���������Ǹ�����ͬ�����ͳ��ֵĸ���,�����㲻ȷ���ԡ�

��ͼ����������Ե��ϵ�����Ļ���������ͼ��
A������о�Ŀ�������Ͳο�ģ��,
B��Ŀ�������Ͳο�ģ��֮�乹��������(pre-phasing),
C����Ŀ�������Ͳο�ģ��֮�乲���ĵ����ͽ��л��������
ʹ�õ�����:һ��������վ,Michigan Imputation Server��վ����:
�����Ļ������������:
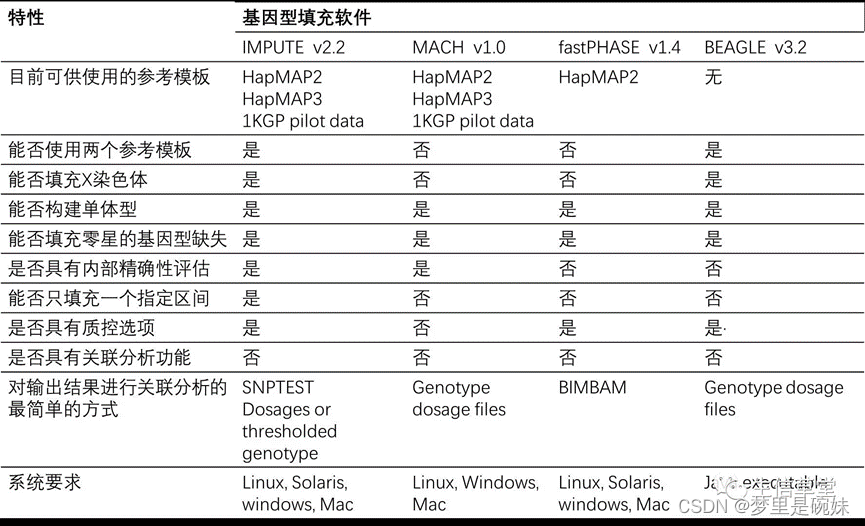
?
3.7Ⱥ��ֲ�У��
GWAS��ͨ������case/control��֮��IJ�����Ѱ���뼲��������SNPλ��,Ȼ��case��control����֮��,���ܱ����ʹ���һ������,��Ӱ����������ļ�⡣
Ⱥ��ֲ�(Population stratification),������IJ�����Դ,ָ����case/control������������ڲ�ͬ������Ⱥ��,����ͽ����Ȼ���в���ġ�
Ҳ����˵��:��ͬȺ��SNPƵ�ʲ�һ��,���º���������������ʱ����ܳ��ּ�����λ��(��һ���������ź�λ����ñ����й�,��������Ⱥ��SNPƵ�ʲ����й�),���������Ҫ�ڹ�������ǰ��Ⱥ��ֲ�У����
GWAS������Ŀ����Ѱ�����ڼ������µIJ���,�����IJ��춼����ϵͳ���,�ڽ��з���ʱ,��Ҫ����У����
����Ⱥ��ֲ��У��,ͨ�������ɷַ����ķ���,��PCA,��Ӧ�����·�����nature genetics��,��������:https://www.nature.com/articles/ng1847
��������:plink��GCTA,�����ﲻ��ʵ����ʾ��
4��GWAS������������
������GWAS�г��õķ�����ͼ��,����Ҫ��������������ͼ��QQ plot��

?
?��������ͼ��QQ plot ����R����qqman��,��Լ���㡣
5��GWAS����meta����
Ⱥ��ֲ���GWAS�о���һ���Ƚϳ����ļ�������Դ��Ҳ����˵,������ݴ���Ⱥ��ֲ�,ȴ�����Կ���,��ô�����õ�һ�Ѽ�����λ�㡣��Ⱥ����ֲַ�ʱ,�����ֶξ��ǽ��ֲ��Ⱥ���������,�������meta������ǰ���Ⱥ��ֲ�У����meta������ǰ�ᡣ
�������õ�����:
METAL (fixed-effect model) (https://genome.sph.umich.edu/wiki/METAL_Documentation)
METASOFT (random-effect model) (http://genetics.cs.ucla.edu/meta/)
6��GWAS������������
�ڵõ���������ͼ������������һ���������źż�����һ��
����rs121������ɨ������ij������������ص�λ��(P=1.351e-36),rs124β�����(6.673e-22),Ҳ����ñ����������,��ô���rs124λ���������ñ����������,������Ϊrs124��rs121�߶�������ƽ��(linkage disequilibrium),�������ǿ����Ľ����:rs124��Զ������rs121ˮ�Ǵ��ߡ�Ϊ�˽���������,���Ǿ���Ҫ������������
һ�������ֽ����
(1)���������Ժ�,rs124��������;�������˵��rs124����Ϊrs121�ŵ����ź�������
(2)���������Ժ�,rs124��������;�������˵��rs124ȷʵ��������,��rs121û�й�ϵ��
����:plink
7��GWAS����gene-based��������
gene-based����������SNP-based����������һ�����䡣
��ͳ�����ϵ�GWAS��Ե���SNPλ����з���,��Ѱ���뼲��������״�������SNPλ�㡣
Ȼ��SNPˮƽ��GWAS������������һЩ����,ͨ����������Ǹ��ݾ�����ֵ,����1X10^-6,5X10^-8��ɸѡͳ��ѧ������SNPλ��,��������������˵��ܶ�pֵ����С,������ЧӦ�����Ļ������ڸ��Ӽ�������,���л��������Ǻܶ����Ч�ý�����Ч����,������ɸѡ��ʽ��©���ܶ���Ҫ����Ϣ��
���,��Ҫ�����ڸ���ˮƽ����SNP GWAS�����Ľ��,�����GWAS�����Ч��,һ����gene-based��gene-set-based����ˮƽ��
- gene-based:�ۺϿ���ij�������϶��SNPλ��Ĺ����������,������û����뼲���Ĺ����ԡ�һ�������Ԫ���Իع�ģ��,���ȶ�ij������������SNPλ��Ļ����;������PCA����,��ѡ���еļ������ɷ���Ϊ�ع�������Ա���,ͨ�����Իع��������뼲���Ĺ�����;
- geneset-based:����ˮƽ,Ҳ���Գ�֮Ϊpathwayˮƽ,�ǻ���ˮƽ�Ľ���,�����˻�������ѧ���ܵĹ�����,վ������ѧ���ܵĽǶ����о�ͻ���뼲���Ĺ����ԡ�����self-contained�ķ���,����ͨ��һ�����ɵ���ֵ,����0.05��ɸѡij�������ϵĺ�ѡSNPλ��,�ں�ѡSNPλ��Ļ�����,��һ��ͨ���ϸ����ֵ���������,����5X10-8, ����Ϊ�����ͷ��������ࡣȻ�����Alleles����genotypes, ѡ���Ӧ��ģ������������ԡ�
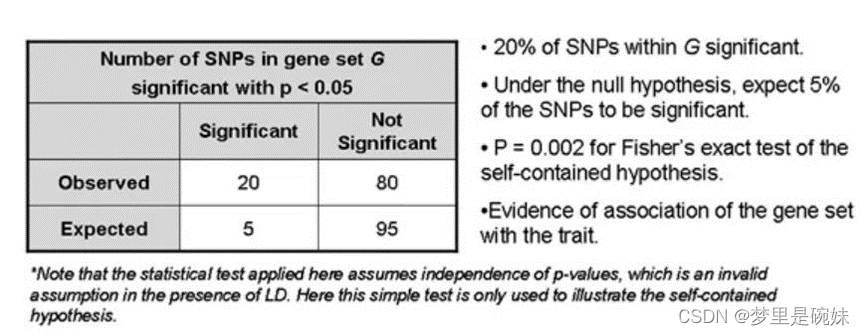
�����ַ����ǽ�����SNP GWASˮƽ�Ļ����ϵ�,�����������Ƕ����е�GWAS����������ж��η���,�����ھ��µ���Ϣ��
����:MAGMA(https://ctg.cncr.nl/software/magma)
8��GWAS�����ϵ¶����������
Katan1986 ���:��ͬ�����;�����ͬ���м����,���ñ��ʹ��������ij��¶����,�û����ͺͼ����Ĺ���ЧӦ�ܹ�������¶���ضԼ���������,���ڵ�λ������ѭ�������ԭ��,�����ò��ܴ�ͳ���в�ѧ�о��еĻ������غͷ������������Ӱ�� (�����������,ָ���DZ�¶�ͽ�ֵ�ʱ��˳��ߵ�)��
?�����о�֤ʵ,�������������ݵ�����ˮƽ,�������;����м���Ͳ���,���м���ʹ���ij��¶���������ڸü�����

?
9��GWAS����LocusZoomͼ
LocusZoomͼ������GWAS���µıر�ͼ��֮һ,����Ҫ�����ǿ��Կ��ٿ��ӻ�GWAS�ҳ������ź��ڻ�����ľ�����Ϣ:������Χ��û�и߶�������λ��,�߶�������λ���Ƿ�Ҳ������
������locuszoom��ʾ��ͼ:
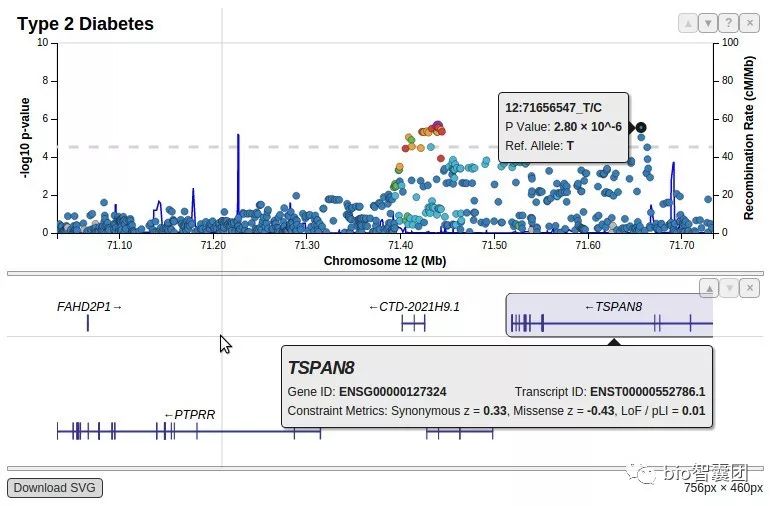
��ƪblog��ʱ�ܽ�����,�ڴ���һƪ�ܹ�������һ����ͺ�ʵ��GWAS?