
药物和蛋白质(靶)之间有着结合亲和力,如果单独的把药物和蛋白质看做俩个独立的部分而忽略掉它们之间的相互作用以及3D坐标(矩阵S)的话是不合理的(如图b),因此本文提出将二者结合成为一个graph(图C)
,称为SPoG,其原子间距离矩阵为D(因为有各个原子的3D坐标S,因此可以转换成原子间的距离矩阵D,但是最主要用到的是距离embedding P),因此问题转化为通过结合图 以及原子距离举证标D来预测药物靶向结合亲和力(Drug-Target Binding Affinity)
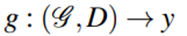
所提出的SpoG结构 
,其中V为蛋白质和药物结点的并集,由于药物和蛋白质之间没有天然的键连接,因此需要二者之间的边,如下:

其中 为设立的threshold,当俩个原子之间的距离小于该值则认为在SpoG图中存在一条边
为设立的threshold,当俩个原子之间的距离小于该值则认为在SpoG图中存在一条边

为了利用拓扑结构和坐标信息,本文提出了S-MAN,里面有一个分层的注意力聚合结构:edge-level aggregation → node-level aggregation,其中采用边级聚合来传递具有位置信息的成对原子embedding以获得边embedding,之后的结点聚合可以获得每个原子结点的边embedding的distance-aware attention,获得边embedding如下公式:
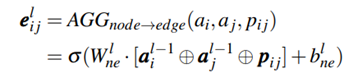
关于结点聚合,首先需要将边embedding以及结点embedding转换为相同的vector,如下:
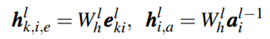
之后结合结点、边、位置信息得到注意力分数

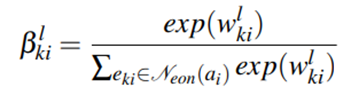
- 看论文代码的数据预处理部分,主要是先得到药物与配体的feature、坐标、边集合、预测值,其中不考虑H原子与其他原子之间的连接及其坐标
1.1 然后根据药物与配体各自的坐标得到药物与配体原子两两之间的距离
1.2 若药物、配体原子俩俩之间的距离小于设定值,认为这俩原子之间存在一条边,将配体原子映射到新的index,设为node_map, 得到药物-配体间的相互作用力图
1.3 构造单独的药物图、配体图
1.4 若药物原子与药物原子之间的距离小于设定值,默认也存在一条相互作用力边,构造该药物空间图的边,同理,适用于配体,得到配体空间图的边,不过在这里要对配体的空间图的边进行特殊处理,用上面得到的node_map值作为新的边
1.5 输出size (药物原子数+配体原子数),feature(药物原子feature和配体原子feature按行拼凑),edges(药物原子空间边,配体原子空间边,药物-配体原子相互作用力边),coords(药物原子左边和配体原子坐标按行进行拼凑)
总结:经过上述处理对数据进行预处理,保存到新的文件中,以后读取数据都从这个新的文件中进行读取,不过在这里貌似没有考虑原子之间本来就存在的边,有点疑惑
继续看了dataset(继承自己定义的BaseDataset)处理过程
3.1 从之前预处理的文件中得到对应的size(num_node)、feature、edges、coords
3.2 根据num_node、feature、edges构造e2n_graph(没有很看明白在干什么,暂时 只有猜测)
3.3 构造e2e_graph,具体意义为如果k-j有条边,j-i有条边,那么在e2e_graph中,
k-j边旁边的节点应该是j-i边,构造e2e_graph的目的就在于此
3.4 空间feature计算:算出edges的左右俩边原子之间的距离dist,然后进行强制截断,距离小于1的强制设为0,距离大于4的强制设置为4,并且强制将原子间距离设为int,然后将原子间的坐标用one-hot向量表示,例如3表示为[0,0,1,0]
3.5. 用pgl构造e2n_graph与e2e_graph图,注入相对应的feature,然后一起append到一个graph_list中
3.6 其get_item方法每次返回graph_list中的一个值、目标值、当前图对应的edges、空间feature、节点数、边数