MolCLR:һ�����ڷ��ӱ���ѧϰ���Լල���
����:
Yuyang Wang1,2, Jianren Wang3 , Zhonglin Cao1 , and Amir Barati Farimani1,2,4,*
1Department of Mechanical Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA
2Machine Learning Department, Carnegie Mellon University, Pittsburgh, PA 15213, USA
3Robotics Institute, Carnegie Mellon University, Pittsburgh, PA 15213, USA
4Department of Chemical Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA
*corresponding author: Amir Barati Farimani (barati@cmu.edu)
ժҪ ABSTRACT
���ӻ���ѧϰ(ML)�ڷ�������Ԥ���ҩ��ַ��������Ҫ��Ӧ��ǰ����Ȼ��,��ȡ��Ƿ��������ǰ���ͺ�ʱ�ġ����ڱ�ע��������,�ලѧϰ��MLģ��Ҫ�ƹ㵽��Ļ�ѧ�ռ���һ�������ս�����������,���������MolCLR:ͨ��ͼ������(GNNs)�� ���� ���з��ӶԱ�ѧϰ(Molecular contrast Learning of representation),����һ����������δ�������(10MΨһ����)�����Ҽලѧϰ����Լලѧϰ | Self-supervised Learning����MolCLRԤѵ����,���ǽ�������ͼ�Ϳ���GNN��������ѧϰ �����ֵı������������������ͼ���㷽��:ԭ���ڱΡ���ɾ������ͼɾ��������ķ���ͼ��ǿ������֤����ǿʱͬһ���ӵ�һ��������Լ���ͬ����һ���Ե���С����ʵ�����,���ǵĶԱ�ѧϰ������������gnn�ڲ�ͬ�������ʻ��ϵ�����,��������ͻع��������ڶԴ���δ������ݿ��Ԥѵ��,��������,MolCLR�����ڼ���������ս�ԵĻ������дﵽ�����Ƚ���ˮƽ������,��һ�����о�����,MolCLRѧ���˽�����Ƕ�������,�����ֻ�ѧ�Ϻ����ķ��������ԡ�
���� Introduction
�ڹ��ܻ���������ͻ�����������,���ӱ����ǻ����ͱز����ٵġ����ڿ��ܵ��ȶ�������������,����һ����Ϣ�ḻ�ı���ģ��������������ѧ�ռ���һ������ս����ͳ�ķ��ӱ���,����չ������ָ��(ECFP)�Ѿ���Ϊ���㻯ѧ�ı����ߡ�����o���Ż���ѧϰ�����ķ�չ�o���������ķ��ӱ���ѧϰ����Ӧ��Խ��Խ������(�绯ѧ����Ԥ�⡢��ѧ��ģ���ͷ������)��
Ȼ��,��������������Ҫ����ս�o����ѧϰ�����ı���������:
-
���ѳ��ױ��������Ϣ��
��������ַ����ı�ʾ��,��SMILES��SELFIES,����ֱ�ӱ�����Ҫ��������Ϣ,Ϊ�˱����ḻ�Ľṹ��Ϣ,��������������ͼ������(GNNs)��
-
��ѧ�ռ�Ĺ�ģ�Ǿ�ġ�
����,DZ�ڵ�ҩ�����Է��ӵĴ�С��������������
����κη��ӱ�����DZ�ڵĻ������н��и��������˺ܴ�����ѡ�
-
����ѧϰ����ı�ǩ���ݺܰ���,��ԶԶ�����o�ر�����DZ�ڵĻ�ѧ�ռ�Ĵ�С���,��÷������Եı�ǩͨ����Ҫ����ijɱ���
���������ѧϰ���еı�ǩ������ԶԶ������,����������������ѵ���Ļ���ѧϰģ�ͺ�������ϡ�
���Ż���ѧϰ�ر������������(DNNs)�ķ�չ�ͳɹ�,���ӱ�ʾѧϰ�ڹ�ȥ��ʮ����Ѹ�ٷ�չ���ڴ�ͳ�Ļ�ѧ��Ϣѧ��,�����Զ��ص�ָ��(FP)�����ʾ,��ECFP�����ǵ�FPs, DNNs������Ԥ��ijЩ���Եġ�����FP��,�����ַ����ı�ʾ(����SMILES)Ҳ���㷺Ӧ���ڷ���ѧϰ��������RNNs�����ϵ�����ģ��ֱ���ʺ�smiles32,33��ѧϰ��ʾ�������������transform�ļܹ��ijɹ�,����������ģ��Ҳ������ѧϰ����SMILES�ı�ʾ��MPNN��D-MPNNʵ���˴ӷ���ͼ�оۺ���Ϣ����Ϣ������ϵ�ṹ������,SchNetģ����GNN�з���֮�����������á�DimeNet����ԭ�Ӽ�ĽǶȽ�����Ϣת��,ʵ�ַ�����Ϣ�ļ��ɡ�
�����ڿ��÷������ݵ�����,�Լල/Ԥѵ�����ӱ���ѧϰҲ���о�����BERT���������Ҽල����ģ���Ѿ�������ѧϰ��SMILES��Ϊ����ķ��ӱ�ʾ���ڷ���ͼ��,N-Gram Graphͨ���̲���װ����Ƕ��������ͼ�ı�ʾ,����ѵ����Hu��������˽ڵ㼶��ͼ����GNNԤѵ������Ȼ��,ͼ����Ԥѵ���ǻ��ڼලѧϰ����,���������ޱ�ǩ��Լ�������˽��Ա�ѧϰ��չ���ǽṹ��ͼ����,���ÿ�ܲ�����ר��Ϊ����ͼѧϰ��Ƶ�,ֻ�������ķ��������Ͻ���ѵ����
���������MolCLRͨ��ͼ������Ա������з��ӶԱ�ѧϰ,�Խ��������ս��MolCLR��һ���Լල��ѧϰ���,�����ھ���1ǧ������ط��ӵĴ����ޱ�Ƿ������ݼ���ѵ�������ġ�ͨ���Ա���ʧ,MolCLRͨ���ԱȻ����ķ���ͼ�Ժ������ķ���ͼ����ѧϰ��ʾ�����������ַ���ͼ����ǿ����:ԭ�����Ρ���ɾ������ͼɾ�����㷺ʹ�õ�GNNģ�͡�ͼ��������(GCN)��ͼͬ������(GIN),������ΪMolCLR�е�GNN������,�Դӷ���ͼ����ȡ��Ϣ��ʾ��Ȼ����MoleculeNet�����η�������Ԥ����϶�Ԥѵ����ģ�ͽ���������ͨ���ලѧϰѵ����GCN��GIN���,���������MolCLR��������˷���ͻع���������ܡ������ڶԴ������ݿ��Ԥѵ��,MolCLR�ڶ�����ӻ������г����������Լලѧϰ��Ԥѵ�����ԡ�����,��һЩ������,MolCLR������ලѧϰ�Ļ���������,���������˼ලѧϰ,���а������ӵķ���ͼ�����������ض��������������MolCLR��ʾ���ʹ�ͳ��FPs֮��Ľ�һ���Ƚϱ���,MolCLR����ͨ���Դ���δ������ݵ�Ԥѵ����ѧϰ���ַ��ӵ������ԡ�
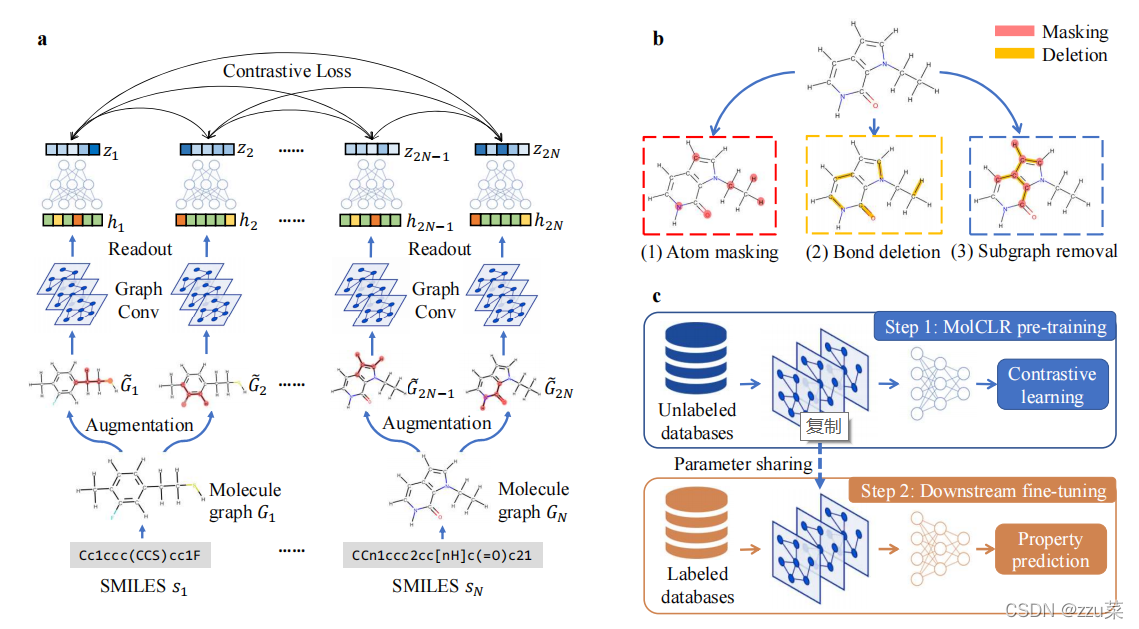
Figure 1.MolCLR�ĸ�����
(a) MolCLRԤѵ��:��С����N�����������е�SMILES
S
n
S_n
Sn?ת��Ϊ����ͼ
G
n
G_n
Gn?����ÿ��ͼӦ�������������ͼ�����������,�õ�������ص��ڱ�ͼ:
G
2
n
?
1
G_{2n-1}
G2n?1?��
G
2
n
G_{2n}
G2n?��һ����������������������ͼ�ξ����Ͷ��������Ļ�����,��ȡ��������
h
2
n
?
1
h_{2n-1}
h2n?1?,
h
2
n
h_{2n}
h2n?�����öԱ���ʧ�����MLP project head��DZ������
z
2
n
?
1
z_{2n-1}
z2n?1?��
z
2
n
z_{2n}
z2n?֮���һ���ԡ�
(b)����ͼ��ǿ����:ԭ���ڱΡ���ɾ������ͼɾ����
?����MolCLR���:����ͨ��MolCLR��GNNs����Ԥѵ��,ѧϰ���������������η�������Ԥ���������GNN��������Ԥѵ������,�������ʼ��һ��MLPͷ��Ȼ����ѭ�ලѧϰ��ѵ��ģ�͡�
��������,(1)���������һ�����ڷ��ӱ�ʾѧϰ���Լලѧϰ���MolCLR��(2)����3�ַ���ͼ��ǿ�������ɶԱȶ�,��ԭ���ڱΡ���ɾ������ͼɾ����(3)�����ڶԴ���δ������ݵ�Ԥѵ��,ͨ��MolCLRѵ���ļ�GNNģ����ලѧϰ���,�����з��ӻ��϶��������ĸĽ���(4)��������δ������ݵĸ����ӵ�GNNģ�����,MolCLR�����ڼ������ӻ���ͨ��������GNNģ�����������Ƚ���(SOTA)ˮƽ
Results
MolCLR Framework

���ǵ�MolCLRģ���ǽ����ڶԱ�ѧϰ���֮�ϵġ����������ͼ�Ե�DZ�ڱ�ʾ�븺�Եı�ʾ�����˶Աȡ�����pipline(ͼ1(a))���ĸ��������:���ݴ�������ǿ������GNN��������ȡ����������project head��һ��temperature-scaled������(NT-Xent)�Ա���ʧ��
������СΪN��mini-batch SMILES���� s n s_n sn?,������Ӧ�ķ���ͼ G n G_n Gn?,����ÿ���ڵ����һ��ԭ��,ÿ���ߴ���ԭ�Ӽ�Ļ�ѧ�������÷���ͼ�������,�� G n G_n Gn?ת��Ϊ������ͬ����صķ���ͼ: G i G_i Gi?�� G j G_j Gj?,���� i = 2 n ? 1 , j = 2 n i = 2n-1, j = 2n i=2n?1,j=2n����ͬһ������������ķ���ͼ��ʾΪ����,���ɲ�ͬ������������ķ���ͼ��ʾΪ���ԡ�������ȡ�� f ( ? ) f(��) f(?)��GNNs��ģ,��������ͼӳ�䵽��ʾ h i , h j �� R d h_i,h_j \varepsilon\mathbb{R}^d hi?,hj?��Rd�С������ǵ�������,������ƽ������Ϊ������ȡ����ʵ��GCN��GIN��������project head g ( ? ) g(��) g(?)���ô������MLPģ��,����ʾ h i h_i hi?�� h j h_j hj?�ֱ�ӳ��Ϊ������ z i z_i zi?�� z j z_j zj?������һ��temperature ������(NT-Xent) lossӦ����2N��DZ����z s,ʹ���Ե�һ�������,���Ե�һ������С���������������PubChem40��10Mδ��������Ͻ���Ԥѵ���ġ�

����MolCLRԤѵ����GNNģ�ͱ������ڷ�������Ԥ��,��ͼ1?��ʾ����ѵ��ǰģ������,Ԥ��ģ����һ��GNN�Ǹɺ�һ��MLP head���,ǰ����ѵ��ǰ��������ȡ��ʹ����ͬ��ģ��,���߽�����ӳ�䵽Ԥ��ķ��������С�ͨ��Ԥѵ��ģ�͵IJ�����������ģ���е�GNN�Ǹɽ��г�ʼ��,����MLPͷ���������ʼ����Ȼ����Ŀ������������ݿ����Լලѧϰ�ķ�ʽѵ��������ģ�͡�����ϸ�ڿ����ڷ���һ�����ҵ���
Molecule Graph Augmentation |����ͼ��ǿ

We employ three molecule graph data augmentation strategies (Figure 1(b)) for input molecules in MolCLR: atom masking,bond deletion, and subgraph removal.
���Ƕ����������MolCLR�в��������ַ���ͼ������ǿ����(ͼ1(b)):ԭ���ڱΡ���ɾ������ͼɾ����
ԭ������(Atom Masking)����ͼ�е�ԭ���Ը����ı�����������Ρ���һ��ԭ�ӱ��ڱ�ʱ,����ԭ��������һ�������� ȡ��,�ñ����ͼ1 b�к����ʾ�ķ���ͼ�е��κ�ԭ������������ͨ������,ģ�ͱ���ѧϰ���ڵĻ�ѧ��Ϣ(������ijЩ�������ӵ�ԭ�ӵĿ������͡�
**��ɾ��(Bond Deletion)**��ͼ1(b)�еĻ�ɫ������ʾ, "��ɾ��"��һ���������ɾ��ԭ��֮��Ļ�ѧ������ԭ�����η����������������ԭʼ������ԭ�����벻ͬ,��ɾ����һ�ָ��ϸ����ǿ,��Ϊ���ӷ���ͼ����ȫɾ���˱�Ե��ԭ��֮�仯ѧ�����γɺͶ��Ѿ����˻�ѧ��Ӧ�з��ӵ����ԡ�����ɾ��ģ���˻�ѧ���Ķ���,��ʹģ��ѧϰһ�������ڸ��ַ�Ӧ�еĹ����ԡ�
**��ͼɾ��(Subgraph Removal)**��ͼɾ�����Ա���Ϊ��ԭ�����κͼ�ɾ���Ľ�ϡ���ͼ��ȥ����һ�������ѡ��ԭ�㿪ʼ��ȥ������ͨ������ԭԭ�ӵ�����,Ȼ�������������,ֱ�������ε�ԭ�������ﵽԭ��������һ��������Ȼ��,�����ε�ԭ��֮��Ļ�ѧ����ɾ��,���������ε�ԭ�Ӻͱ�ɾ���Ļ�ѧ�����γ���ԭ����ͼ����ͼ����ͼ1(b)�е���ɫ������ʾ,���Ƴ�����ͼ�������ڱ�ԭ��֮������л�ѧ����ͨ��ƥ�䱻�Ƴ��IJ�ͬ�ӽṹ�ķ���ͼ,��ģ��ѧ������ʣ�����ͼ���ҵ���������,���ںܴ�̶��Ͼ����˷��ӵ����ԡ�
Molecular Property Predictions | �������ʵ�Ԥ��

Table 1. Test performance of different models on seven classifification benchmarks. The fifirst seven models are supervised learning methods and the last four are self-supervised/pre-training methods. Mean and standard deviation of test ROC-AUC (%) on each benchmark are reported.
*Best performing supervised and self-supervised/pre-training methods for each benchmark are marked as bold.
��1�����߸�������ϲ��Բ�ͬģ�͵����ܡ�ǰ�߸�ģ�����мල��ѧϰ����,���ĸ������Ҽල/ѵ��ǰ�ķ���������ÿ��������ROC-AUC(%)��ƽ��ֵ�ͱ�ƫ�
**����������ѵļල�����Ҽල/ѵ��ǰ�����Դ����ǡ�

Table 2. Test performance of different models on six regression benchmarks. The fifirst seven models are supervised learning methods and the last four are self-supervised/pre-training methods. Mean and standard deviation of test RMSE (for FreeSolv,ESOL, Lipo) or MAE (for QM7, QM8, QM9) are reported.
*Best performing supervised and self-supervised/pre-training methods for each benchmark are marked as bold.
��2���������ع���ϲ��Բ�ͬģ�͵����ܡ�ǰ�߸�ģ�����мල��ѧϰ����,���ĸ������Ҽල/ѵ��ǰ�ķ����������˲���RMSE(����FreeSolv, ESOL, Lipo)��MAE(����QM7, QM8, QM9)��ƽ��ֵ�ͱ��
- *����������ѵļල�����Ҽල/ѵ��ǰ�����Դ����ǡ�
Ϊ��֤��MolCLR����Ч��,���Ƕ�����MoleculeNet�Ķ��������ս�Եķ���ͻع���������ܽ����˻����ԡ��������ݼ�����ϸ��Ϣ�������1�Ͳ����2����1��ʾ����ල�����Ҽල/ѵ��ǰ����ģ�����,���ǵ�MolCLRģ���ڷ��������ϵIJ���ROC-AUC(%)�������������������е�ƽ���ͱ�ƫ�MolCLRGCN��MolCLRGIN�ֱ��ʾ��GCN��GIN��Ϊ������ȡ����MolCLRԤѵ������1�Ĺ۲�������:
- �������Լලѧϰ��Ԥѵ���������,���ǵ�MolCLR�����7������������5���ﵽ���������,ƽ�������4.0%�������ĸĽ�˵�������ǵ�MolCLR��һ��ǿ������Ҽලѧϰ����,��������ʵ��,���Ҷ��ض�����ĸ�����Ҫ����١�
- �������õļලѧϰ�������,MolCLRҲ���ֳ��˾������ܡ���һЩ��������(����,ClinTox, BACE, MUV),���ǵ�ѵ��ǰģ������������SOTA�ලѧϰ����,���а������ӵľۺϲ������ض����������������,��ClinTox��,���мල��D-MPNN���,MolCLRʹROC-AUC�����2.7%��
- ֵ��ע�����,MolCLR�����������ķ������ݼ��ϱ��ֵ÷dz���,��ClinTox��BACE��SIDER����������֤��MolCLRѧϰ��Ϣ��ʾ,�����ڲ�ͬ�����ݼ�֮�䴫�䡣
��2������MolCLR�ͻ���ģ���ڻع���ϵIJ������ܡ�FreeSolv��ESOL��Lipo���þ��������(RMSE)��Ϊ����ָ��,��QM7��QM8��QM9���þ������������(MAE)���в���,����ѭMoleculeNet�Ľ��顣��������,�ع����������ս��,��Ϊ����ֻ�����ֹ��������ɢ��ǩ����2�Ĺ۲������¡�
- MolCLR��6��������5������������ѵ��ǰ��,�������ESOL����ȡ���˼�����ͬ�����ܡ���Hu����45ͬ��ʵ��GIN��Ϊ���������,MolCLRGIN������6���ع����ݿ��ϵ����ܶ�����Hu����45����QM7��QM9Ϊ��,��Hu�ȵĸĽ��ʷֱ�Ϊ20.9%��45.8%��
- ��ලѧϰģ�����,MolCLR�ڴ��������´ﵽ�������ܡ�����,��Lipo���ݿ���,MolCLR�õ�����������õļලD-MPNN���ƵĽ��������,GCN��GINͨ�������лع������MolCLRԤѵ������˸��õ�Ԥ�����ܡ���Ȼ,��QM9��,MolCLR������ල��SchNet19��MGCN52��������Ϊ������ģ����ר��Ϊ���ӽ���������Ƶ�,���������˶����3Dλ����Ϣ��ֵ��ע�����,����SchNet��MGCN���漰������ѧ���Ե����ݼ�(��QM7��QM8��QM9)�ϱ��ֳ�����Խ������,������������,���Dz�û����ʾ�����������ලѧϰ���ߵ����ơ�����,�ھ�����ս�Ե�QM9��������,MolCLRԤѵ����Ȼ��֤������Ч�ġ���δ����ѵ����GCN��GIN���,MolCLR���������������,�ֱ����38.7%��50.3%��ͬʱ,��QM9��,MolCLR��ѧϰЧ�����������Լලѧϰ����,��֤��MolCLR����Ч�ԡ�����QM9�����ʾ��в�ͬ�ĵ�λ������,QM9����ϸ��������ڲ����3�С�
�ӱ�1�ͱ�2���Կ���,��ල��GCN��GIN���,MolCLRԤѵ�������л��ϵ����ܶ��кܴ�����,��˵����MolCLR����Ч�ԡ��ڷ������,ͨ��MolCLR��ƽ������,GCNΪ12.4%,GINΪ16.8%��ͬ��,�ڻع������,GCN��GCN��ƽ�������ʷֱ�Ϊ27.6%��33.5%���ܵ���˵,ͨ��MolCLRǰѵ��,GIN��GCN���ֳ�����Ľ��������������ΪGIN�и���IJ���,�ܹ�ѧϰ���д����Եķ�������������,�ڴ���������,������ѵ��ǰ/���Ҽලѧϰ�������,MolCLR��ʾ�����õ�Ԥ�⾫�ȡ���Ҫǿ������,MolCLR�����ڶԴ���δ������ݿ��Ԥѵ��,�������ල/���Ҽලѧϰ������Ȼ������δ�������ΪMolCLR�ڻ�ѧ�ռ���ַ�������֮��ķ����ṩ�˱��������߸�������ơ������4�Ͳ���ͼ1��һ���о���ѵ��ǰ���ݿ��MolCLR��Ӱ�졣���ַ�������ΪԤ��ҩ��ֺ������DZ�ڵķ��������ṩ��ϣ����
Optimal Molecule Graph Augmentations | ���ŷ���ͼ��ǿ
Ϊ��ϵͳ�ط�������ͼ��ǿ���Ե�Ч��,���DZȽ���ԭ���ڱΡ���ɾ������ͼɾ���IJ�ͬ��ɡ�ͼ2(a)��ʾΪÿ��������ǿ�����ڲ�ͬ���ϵ�ROC-AUC(%)ƽ��ֵ�ͱ��������������ǿ���ԡ�(1)ԭ���ڱκͼ�ȱʧ������,���ߵı���p����Ϊ25%��(2)�������pΪ0% ~ 25%����ͼɾ����(3)�Թ̶���25%����ȥ����ͼ��(4)�������㷽������ϡ�������˵,����Ӧ���������Ϊ0%��25%����ͼɾ����Ȼ��,�����ģԭ�ӵı���С��25%,���Ǽ��������ģԭ��,ֱ���ﵽ25%�ı�����ͬ��,�����ɾ����С��25%,��ɾ������ļ�,�Դﵽ�趨�ı��ʡ�
��ͼ2(a)��ʾ,�����������,25%����ͼɾ����ƽ���ﵽ��õ����ܡ������(1)���,ȥ����ͼ��ԭ���ڱκͼ�ɾ�������ڽ��,����ȥ����ͼ��һ�������˾ֲ��ӽṹ,���,���㷨���������ܿɹ����ڴˡ���BBBP���ݼ���,�̶�25%��������ͼɾ��Ч���ϲ�,������ΪBBBP�еķ��ӽṹ�Ƚ�����,���˽ṹ��С�仯�ᵼ�����ʵľ���졣����,ֵ��ע�����,�ڴ������������,�뵥��ͼɾ����ǿ���,��������ǿ(����(4))����ϻ���ROC-AUC��һ�����ܵ�ԭ����,��������ǿ���Ե���Ͽ���ȥ������ͼ�й㷺���ӽṹ,�Ӷ�������Ҫ��������Ϣ��һ����˵,��ͼɾ���ڴ�����������ж��ܻ�ø��õ����ܡ�Ȼ��,��Ҳ����,���ŵķ���ͼ��������������ġ�

Figure 2. Investigation of molecule graph augmentations on classifification benchmarks. (a) Test performance of MolCLR models with different compositions of molecule graph augmentation strategies. (b) Test performance of GIN models trained via supervised learning with and without molecular graph augmentations. The height of each bar represents the mean ROC-AUC(%) on the benchmark, and the length of each error bar represents the standard deviation.
ͼ2.���ڷ�����ķ���ͼ��ǿ���о���
(a)��ͬ����ͼ��ǿ������ɵ�MolCLRģ�͵IJ������ܡ�
(b)����ͨ���к�û�з���ͼ��ǿ�ļලѧϰѵ����GINģ�͵����ܡ�ÿ��������ĸ߶ȴ������ϵ�ROC-AUC(%)��ƽ��ֵ,ÿ��������ij��ȴ������
Molecule Graph Augmentation on Supervised Learning | �мලѧϰ�ķ���ͼ��ǿ
���ǹ����еķ���ͼ��չ����,��ԭ���ڱΡ���ɾ������ͼɾ��,������Ϊһ��ͨ�õ�������ǿ�����ʵ��,�������κλ���ͼ�ķ���ѧϰ������Ϊ����֤����ͼ��ǿ�Լල�����������Ч��,���Ǵ������ʼ����ʼѵ������ǿ��û����ǿ��GINģ�͡�������˵,ʵ���˹̶�����Ϊ25%����ͼ�ڱ���Ϊ��ǿ��ͼ2(b)��¼�˲���ROC-AUC(%)������߸��������ʷ������ƽ��ֵ�ͱ���������߸���������,ʹ����ǿѵ����GINs��������û����ǿѵ����ģ�͡�����ͼ��ǿ�������ƽ��ROC-AUC����7.2%�������ǵķ���ͼ��ǿ����Ӧ�����мල�ķ�������Ԥ������,��ʹû��Ԥ��ѵ��,Ҳ�ܴ��������ܡ��������,����ͼ���������Ч�ذ���gnnѧϰ³���Ժʹ���������������,��ͼɾ��ƥ�䲿�ֹ۲쵽�ķ���ͼ�����,ģ��ѧϰ��ʣ�����ͼ��Ѱ�����ŵĻ���,��Է������ʵ�ѧϰ�dz�������
Investigation of MolCLR Representation | MolCLR�������о�
����ʹ��t-SNE embedding�����Ԥѵ����MolCLRѧϰ�ı�ʾ��t-SNE�㷨�����Ƶķ��ӱ�ʾӳ�䵽��ά(2D)�е����ڵ㡣ͼ3��ʾ��PubChem���ݿ���֤���е�100K����,ͨ��t-SNEǶ�뵽2D��,���ݷ�������ɫ�����ǻ���ͼ�м�����һЩ���ѡ��ķ���,��˵��ͨ��MolCLRԤѵ��ѧϰ��������/�����Ʒ�������Щ����ͼ3��ʾ,���ھ����������˽ṹ�����ŵķ���,MolCLRѧϰ�˽ӽ��ı�ʾ������,������ʾ���������Ӿ����뷼���������ʻ������½���ʾ���������ӽṹ����,����һ��±��ԭ��(������)�뱽��������˵��,��ʹû�б�ǩ,��ģ��Ҳ��ѧϰ����֮���������ϵ,��Ϊ�����������ʵķ��Ӿ�������������������MolCLR��ʾ�Ŀ��ӻ������ڲ���ͼ2���ҵ���
- Embedding���Ǵ�ԭʼ������ȡ������Feature,Ҳ�����Ǹ�ͨ��������ӳ��֮��ĵ�ά������
Ϊ�˽�һ������MolCLR,���ǽ�MolCLRѧϰ�����봫ͳ�ķ���ָ��(FPs)�����˱Ƚ�,��ECFP5��RDKFP���ر��,����һ����ѯ����,����ͨ��MolCLR��ȡ���ʾ,�������������ǵ�ѵ��ǰ���ݿ������вο����ӵ����Ҿ��롣������ʾ ( u , v ) (u, v) (u,v)֮������Ҿ��붨��Ϊ 1 ? u ? v �� u �� �� v �� 1-\frac{u \cdot v}{\|u\|\|v\|} 1?��u����v��u?v?��Ȼ����ݱ�ʾ��������вο����ӽ�������,����������ٷֱȾ��ȵط�Ϊ20��Ͱ���ٷֱ���ֵԽ��,��ѯ�����������Ʒ���Խ��,��ΪMolCLR��ʾԽ�ӽ�����ÿ��bin��,���ѡ��5000������,�������������ѯ��dice FP�����ԡ�ͼ4��ʾ��һ����ѯ���ӵ�����(PubChem ID 42953211)����ͼ4(a)��ʾΪÿ������FP���ƶȵľ�ֵ�ͱ��ʹ��ECFP��RDKFP�����ƶȷֲ���ͼ4(b)��ʾ��ECFP��RDKFP���и��͵�������,��Ϊǰ�ߺ����˸��㷺������ӻ�����ص��������������,����MolCLR��ʾ���������,ECFP��RDKFP�����ƶȶ������ˡ���ǰ5%��,RDKFP��ƽ�����ƶ�Ϊ0.9,���ں��5%��,RDKFP��ƽ�����ƶ�Ϊ0.67��ͬ��,ƽ��ECFP�����Դ�ǰ5%��0.49�½�����5%��0.21����Ȼ���Űٷֱ���ֵ�����ӻ��в���,������������MolCLRѧϰ�����ͻ�ѧָ��(FPs)֮����һ�µġ���MolCLR���ɵı�ʾ ��ľ�����Ч�ط�ӳ�˷���������������,ͼ4?��������MolCLR��ʾ�������ѯ������ӽ���9������,����֡�����ƶȶ���ע�ˡ���Щ���Ӿ��нϸߵ�RDKFP������,��0.833��0.985,��һ��֤����MolCLRѧϰ��ѧ����������۲쵽,��Щѡ��ķ��ӹ�����ͬ�Ĺ�����,�������±����(��),�尷,ͪ,���㻯�����ԽṹҲ���������з������ҵ���ֵ��ע�����,ͼ4?�е�һ�еĵڶ����������ѯ������ȫ��ͬ,�����ȵ�λ��,�����������ߡ��������,ͨ���Դ�����δ������ݽ��жԱ�ѧϰ,MolCLR�ܹ��Զ�������Ƕ�뵽���д����Ե�������,���Ի�ѧ�Ϻ����ķ�ʽ�Ի�����������֡��ڲ���ͼ3�п����ҵ������ѯ���ӵ����ӡ�

Figure 3. Visualization of molecular representations learned by MolCLR via t-SNE. Representations are extracted from the validation set of the pre-training dataset, which contains 100k unique molecules. Each point is colored by its corresponding molecular weight. Some molecules close in the representation domain are also shown.
ͼ3��MolCLRͨ��t-SNEѧϰ�ķ��ӱ����Ŀ��ӻ�����ʾ ��ѵ��ǰ���ݼ�����֤������ȡ,�����ݼ�����100k��Ψһ�ķ��ӡ�ÿ���㶼����Ӧ�ķ�������ɫ��һЩ�����ڱ�ʾ�����е�Ҳ��ʾ��

Figure 4. Comparison of MolCLR-learned representations and conventional FPs using the query molecule (PubChem ID42953211). (a) Change of ECFP and RDKFP similarities with respect to the distance between MolCLR representations. (b)Distribution of ECFP and EDKFP similarities with the query molecule. ? The query molecule and 9 closest molecules in MolCLR representation domain with RDKFP and ECFP similarities labeled.
ͼ4��ʹ�ò�ѯ����(PubChem ID 42953211)�Ƚ�molclrѧϰ��ʾ�ʹ�ͳFPs��
(a) ECFP��RDKFP�����������MolCLR��ʾ�����ı仯��
(b)��ѯ���ӵ�ECFP��EDKFP�����Էֲ���
?��MolCLR��ʾ���б�עRDKFP��ECFP�����ԵIJ�ѯ���Ӻ�9����ӽ��ķ��ӡ�
Conclusion
���������,�����о��˷��ӱ�ʾ�����Ҽලѧϰ��������˵,���������ͨ��gnn�����ַ���ͼ��ǿ����(ԭ���ڱΡ���ɾ������ͼɾ��)�����б�ʾ�ķ��ӶԱ�ѧϰ��ͨ���Ա�������������Ժ���,MolCLRʹ��һ��GNN�Ǹ�ѧϰ��Ϣ��ʾ��ʵ�����,����MolCLRԤѵ����GNNģ���ڸ��ַ��ӻ��϶�ȡ���˺ܴ�ĸĽ�,���Լලѧϰ��ʽѵ����ģ�����,���и��õķ������ܡ�
ͨ��MolCLRѧϰ�ķ��ӱ�ʾ��֤���������������¿�ת�Ƶ���������,�Լ��ڴ�Ļ�ѧ�ռ��ϵķ�����������δ���Ĺ�����,������ֵ���о��ķ�������,�Ľ�GNN�Ǹ�(�������transform��GNN�ܹ�)��������ȡ���õķ��ӱ�ʾ������,�����Ҽලѧϰ���������ͽ���Ҳ����Ȥ���������о��������о���Ա���õ��˽⻯����,������ҩ��ķ��֡�
Methods
Graph Neural Networks | ͼ������
�����ǵĹ�����,һ������ͼ G G G������Ϊ G = ( V , E ) G = (V,E) G=(V,E),���� V V V�� E E E�ֱ��ǽڵ�(ԭ��)�ͱ�(��ѧ��)���ִ�ͼ������(GNNS)��������ۺϲ���,�������½ڵ��ʾ��GNN��k��ڵ������ľۺϸ��¹�����ʽ
a v ( k ) = AGGREGATE ? ( k ) ( { h u ( k ? 1 ) : u �� N ( v ) } ) , h v ( k ) = COMBINE ? ( k ) ( h v ( k ? 1 ) , a v ( k ) ) \boldsymbol{a}_{v}^{(k)}=\operatorname{AGGREGATE}^{(k)}\left(\left\{\boldsymbol{h}_{u}^{(k-1)}: u \in \mathscr{N}(v)\right\}\right), \boldsymbol{h}_{v}^{(k)}=\operatorname{COMBINE}^{(k)}\left(\boldsymbol{h}_{v}^{(k-1)}, \boldsymbol{a}_{v}^{(k)}\right) av(k)?=AGGREGATE(k)({hu(k?1)?:u��N(v)}),hv(k)?=COMBINE(k)(hv(k?1)?,av(k)?)
���� h v ( k ) h_v^{(k)} hv(k)?Ϊ�ڵ�v�ڵ�k�������, h v ( 0 ) h_v^{(0)} hv(0)? �ɽڵ����� x v x_v xv?��ʼ���� N ( v ) N (v) N(v)Ϊ�ڵ�v�������ھӵļ��ϡ�Ϊ�˽�һ����ȡͼ������ h G h_G hG?,����������ͼG�е����нڵ�������������,��ʽ2��ʾ
h G = READOUT ? ( { h u ( k ) : v �� G } ) \boldsymbol{h}_{G}=\operatorname{READOUT}\left(\left\{\boldsymbol{h}_{u}^{(k)}: v \in G\right\}\right) hG?=READOUT({hu(k)?:v��G})
�����ǵĹ�����,���ǻ���GCN��GIN������GNN���������ڽ������Ա任֮ǰ,GCNͨ���ڽڵ㱾�������ڽӽڵ�������һ����ֵ�������ɾۺϺ���ϲ�������GIN������MLP�ͽڵ�������Ȩ��͵ľۺϷ��������߶��Ǽ�ͨ�õ�ͼ�������㡣����,����ʵ���˹㷺ʹ�õ�ƽ��ֵ����Ϊ������
Contrastive Learning | �Ա�ѧϰ
�Ա�ѧϰּ��ͨ���Ա������ݶԺ����ݶ���ѧϰ������SimCLR48֤����ͼ��ĶԱ�ѧϰ���Դ�������ǿ�ʹ�����������л�ü���ĺô���SimCLR����InfoNCE loss,���NT-Xent loss��ʽ3��ʾ:
L i , j = ? log ? exp ? ( sim ? ( z i , z j ) / �� ) �� k = 1 2 N 1 { k �� i } exp ? ( sim ? ( z i , z k ) / �� ) \mathscr{L}_{i, j}=-\log \frac{\exp \left(\operatorname{sim}\left(\boldsymbol{z}_{i}, \boldsymbol{z}_{j}\right) / \tau\right)}{\sum_{k=1}^{2 N} \mathbb{1}\{k \neq i\} \exp \left(\operatorname{sim}\left(\boldsymbol{z}_{i}, \boldsymbol{z}_{k}\right) / \tau\right)} Li,j?=?log��k=12N?1{k��?=i}exp(sim(zi?,zk?)/��)exp(sim(zi?,zj?)/��)?
���� z i z_i zi?�� z j z_j zj?Ϊ�����ݶ���ȡ��DZ����,NΪ������С, s i m ( ? ) sim(��) sim(?)������������֮���������, �� �� ��Ϊ�¶Ȳ����������ǵ�MolCLR��,������ѭNT-Xent��ʧ��GNN����������Ԥѵ��,�����������ƶ�ʵ��Ϊ sim ? ( z i , z j ) = z i T z j �� z i �� 2 �� z j �� 2 \operatorname{sim}\left(z_{i}, z_{j}\right)=\frac{z_{i}^{T} z_{j}}{\left\|z_{i}\right\|_{2}\left\|z_{j}\right\|_{2}} sim(zi?,zj?)=��zi?��2?��zj?��2?ziT?zj??�������5�Ԧ���MolCLRѵ��ǰ�Ľ�һ���о������ܶԱ�ѧϰ����Ѿ���Ӧ�õ���������,�����ǽṹ��ͼ�Ρ�����Ƕ��ͻ����˹滮�����ڷ���ͼ�ĶԱ�ѧϰ��û�н���ȫ����꾡���о���
Datasets
Ԥѵ�����ݼ�������MolCLR��Ԥѵ��,����ʹ������ChemBERTa��PubChem�ռ���1000������ص�δ��Ƿ���SMILES��Ȼ������RDKit��������ͼ����SMILES������ȡ��ѧ�������ڷ���ͼ��,ÿ���ڵ����һ��ԭ��,ÿ���ߴ���һ����ѧ����������95/5�ı��������ѵ��ǰ���ݼ��ָ�Ϊѵ��������֤����
�������ݼ���Ϊ�˲������ǵ�MolCLR��ܵ�����,����ʹ��������MoleculeNet��13�����ݼ�,�ܹ�����44����Ԫ���������24���ع�������Щ�����˶������ķ������ԡ����ڳ�QM9֮����������ݼ�,����ʹ��scaffold���������һ��80/10/10 train/valid/test���,���н�����������볣����������ֲ�ͬ,���ڷ����ǽṹ��֧�ܷ���ʹ��Ԥ�����������ս��,��Ҳ������ʵ��QM9��ѭ����ָ�������Ϊ�������ع�����ʵ�ֽ��бȽϡ�
**ѵ��ϸ�ڡ�**����ͼ�ϵ�ÿ��ԭ�Ӱ�ԭ����������������Ƕ��,ÿ���������ͺͷ���Ƕ�롣���ǽ�ReLU������ΪGNN�Ǹ�,ʵ����һ��5��ͼ����,����ѭHu���˵���,ʹ�ۺ����Ե�������ݡ���ÿ��ͼӦ��ƽ���ػ���Ϊ��������,��ȡ512ά�ķ��ӱ�ʾ��һ���������ز��MLP����ʾӳ�䵽һ��256ά��DZ�ڿռ䡣������˥�� 1 0 ? 5 10^{-5} 10?5��Adam�Ż��������Ż�NT-Xent��ʧ������ѧϰ���ʳ�ʼ��10��epoch֮��, 5 ? 1 0 ? 4 5 *10^{-4} 5?10?4,ִ��һ������ѧϰ˥������512��������Сѵ��ģ��,��ѵ��50��epoch��
���������������,�����ڻ���GNN������ȡ��������һ�������ʼ����MLP���Է�������ͻع�����ֱ�ʵ����Softmax��������ʧ��L1��ʧ����ÿ��������,���Ƕ�Ԥѵ��ģ�ͽ���������100 epoch����,�Եõ����Լ����ܵ�ƽ��ֵ�ͱ������ֻ��ѵ������ѵ��ģ��,������֤����ִ�г����������Ի����ѽ������������ǻ���Pytorch Geometricʵ�ֵġ������6�ṩ�˸������ϸ�ڡ�
Baselines
�мලѧϰģ���������ۺ����������ǵ�MolCLRģ�͵�����,��������ලѧϰ���������˱Ƚϡ�����dz�����ѧϰģ��,�������Է���ָ��Ϊ��������ɭ��(Random Forest, RF)��֧��������(Support Vector machine, SVM)�����������GNNS�������˾��б�Ե������GCN��GIN���оۺϡ�����,���ǻ��������ڼ������ӻ��ϴﵽSOTA��GNNģ��(SchNet19��MGCN52��D-MPNN20)��Ϊ����ʵ�֡���ЩGNN��ר��Ϊ������Ƶġ�����,SchNet��MGCN��ȷ��ģ���˷����ڲ�����������á�
�Լලѧϰģ����Ϊ�˸��õ�֤��MolCLR��ܵ���Ч��,���ǽ�һ��������ѵ��ǰ�����Ҽලѧϰģ����Ϊ���ߡ�Hu��������˷���ͼ�Ľڵ㼶��ͼ��Ԥѵ������Ҫָ������,��Ȼ�ڵ㼶��Ԥѵ���ǻ����Լල��,��ͼ����Ԥѵ���ǻ���ijЩ�������ʱ�ǩ�ļල��N-GramͼҲ��ʵ��,��ֱ��ͨ������ͼ����һ�����յı�ʾ��