学习笔记,仅供参考,有错必纠
Journal:Bioinformatics
Year:2019
Authors:Soufiane Mourragui, Marco Loog, Lodewyk F. A. Wessels
PRECISE: a domain adaptation approach to transfer predictors of drug response from pre-clinical models to tumors
Abstract
Motivation: 细胞系和病人衍生的异种移植(PDXs)已被广泛用于了解癌症的分子基础. 虽然核心生物过程通常是保守的,但这些模型与人类肿瘤相比也显示出重要的差异,阻碍了从临床前模型到人类环境的结果转化. 特别是,利用从pre- clinical模型获得的数据生成的药物反应预测器来预测病人的反应仍然是一项具有挑战性的任务. 由于已经为pre- clinical模型收集了大量的药物反应数据集,而病人的药物反应数据往往缺乏,因此迫切需要一种方法,将药物反应预测器从pre- clinical模型有效地转移到人体环境.
Results: 我们展示了,细胞系和PDXs与人类肿瘤有共同的特征和过程. 我们对这种相似性进行了量化,并表明回归模型不能简单地在细胞系或PDX上训练,然后应用于肿瘤. 我们开发了PRECISE,这是一种基于领域适应性的新方法,它在一个共识的表示中捕获了pre- clinical模型和人类肿瘤之间的共同信息. 采用这种表示方法,我们在pre- clinical数据上训练药物反应的预测器,并应用这些预测器对人类肿瘤进行分层. 结果表明,由此产生的领域变量预测器在pre- clinical领域的预测性能有小幅下降,但重要的是,其可靠地恢复了人类肿瘤上独立生物标志物和其配套药物之间的已知关联.
Availability and implementation: PRECISE and the scripts for running our experiments are available on our GitHub page (https://github.com/NKI-CCB/PRECISE).
Introduction
癌症是一种异质性的疾病,由于体细胞基因组改变的积累而产生. 这些改变在肿瘤之间表现出高度的差异性,导致对治疗的反应不一. 精准医疗试图通过考虑到这种异质性并根据特定肿瘤的特定分子构成进行治疗来提高反应率. 这需要识别生物标志物,以确定将从特定治疗中受益的一组病人,同时避免那些不会受益的病人产生不必要的副作用. 然而,由于各种药物的患者反应数据有限,pre- clinical模式,如细胞系和患者衍生的异种移植(PDXs)已被用来产生大规模的数据集,使基于数据驱动的反应生物标志物的识别的个性化治疗策略得以发展. 更特别的是,数百个pre- clinical模型不仅被广泛地进行了分子特征分析,更重要的是,它们对数百种药物的反应也被记录下来. 这导致了大量的公共资源,包括来自细胞系(GDSC1000,Iorio等人,2016)和PDX模型(NIBR PDXE,Gao等人,2015)的数据.
这些pre- clinical资源可以用来建立药物反应的预测器,然后转移到人类环境中,允许对患者尚未接触的药物进行分层. Geeleher等人通过简单地校正细胞系和肿瘤数据集之间的批次效应,然后直接将细胞系预测器转移到人体环境中(Geeleher等人,2014,2017). 这已经产生了一些有希望的结果:它恢复了成熟的生物标志物,如拉帕替尼敏感性和ERBB2扩增之间的关联. 然而,当直接将预测器从source domain(细胞系)转移到target domain(人类肿瘤)时,我们假设源和目标数据来自于相同的分布. pre- clinical模型和人类肿瘤之间的差异已被广泛研究(Ben-David等人,2017,2018;Gillet等人,2013),最明显的差异包括细胞系和PDXs中没有免疫系统,以及细胞系中没有肿瘤微环境和脉管. One can therefore not assume similarity between the source and target distributions.
Transfer learning旨在解决这个问题(see Pan and Yang, 2010 for a general review). 根据源和目标标签的可用性以及这些源和目标数据集之间的具体关系,转移学习方法可以被归为不同的类别. 由于我们有标记的肿瘤样本非常少,但有很多标记的pre- clinical模型,我们的方法属于被称为过渡性的类别[while this terminology is not widely used in the community, we follow the categorization employed in Pan and Yang (2010)]. 由于特征(即基因)在源域和目标域中是相同的,我们的问题需要一个域适应策略,有时也被称为同质域适应.
如前所述,临床前模型和肿瘤的marginal distributions预计是不同的. 然而,我们假设药物反应在很大程度上是由pre- clinical模型和人类肿瘤之间保守的生物现象决定的. 因此,应该存在一组特征(基因),对于这些特征,药物反应的条件分布在细胞系、PDXs和人类肿瘤之间是可比的. 已经提出了不同的方法来寻找这样的共同空间,这些方法可以分为两大类(Csurka, 2017):
- 第一类方法,即data- centric的方法,可以通过aligning the marginal distributions直接从pre-clinical模型和肿瘤中找到一个共同subspace.
- 第二类的方法,称为subspace-centric,通过降低维度,然后aligning the low-rank representations来进行领域适应性修正(Fernando等人,2013;Gong等人,2012;Gopalan等人,2011).
在第一类中,边际分布是直接对齐的,这表明empirical distribution将充分准确地反映source 和 target samples的real behavior. 例如,如果源数据集中ER阳性样本的比例与目标数据集非常不同,这样的方向将被抛弃,因为它在源和目标之间的差异太大. 显然,这是不可取的,因为它代表了乳腺癌中一个非常重要的变量.
第二类找到了important variations的directions,然后在source和target之间比较这些方向. 这些方法并不直接比较分布,而且较少受到样本量问题和样本选择偏差的影响,因此,我们选择第二类方法.
我们提出了Patient Response Estimation Corrected by Interpolation of Subspace Embeddings (PRECISE),这是一种基于领域适应性的方法,在人类肿瘤与临床前模型共享的过程中训练回归模型. 图1展示了PRECISE的一般工作流程.
我们首先通过线性降维从细胞系、PDXs和人类肿瘤中独立提取因子. 然后,使用一个线性转换,将其中一个pre- clinical模型的因子与人类肿瘤因子进行几何匹配(Fernando等人,2013). 随后我们提取共同因素[principal vectors (PVs)],并定义为受线性转换影响最小的方向(图1A).
在选择最相似的PV后,我们通过在源域(cell line or PDX PVs) 和目标域(human tumor PVs)之间插值计算新的特征空间. 这种插值产生的特征空间允许在所选模型系统和肿瘤之间取得平衡(Gong等人,2012;Gopalan等人,2011). 这种方法虽然使用了canonical angle的概念,但与典型相关分析有明显不同. 事实上,在我们的案例中,样本不是成对的,不能计算交叉相关. 从一组插值空间中,通过优化所选pre- clinical模型的边际分布和投射在这些插值特征上的人类肿瘤数据之间的匹配来获得共识表示. 这些共识特征最后被用来训练一个回归模型,使用来自所选临床前模型的数据. 我们用这个回归模型来预测肿瘤药物反应(图1B).
由于这些特征在临床前模型和人类肿瘤之间是共享的,预计回归模型可以推广到人类肿瘤. 最后,我们使用已知的生物标志物-药物关联(from independent data sources, e.g. mutation status, copy number)作为阳性对照,以验证该模型在人类肿瘤中的预测结果(图1C).
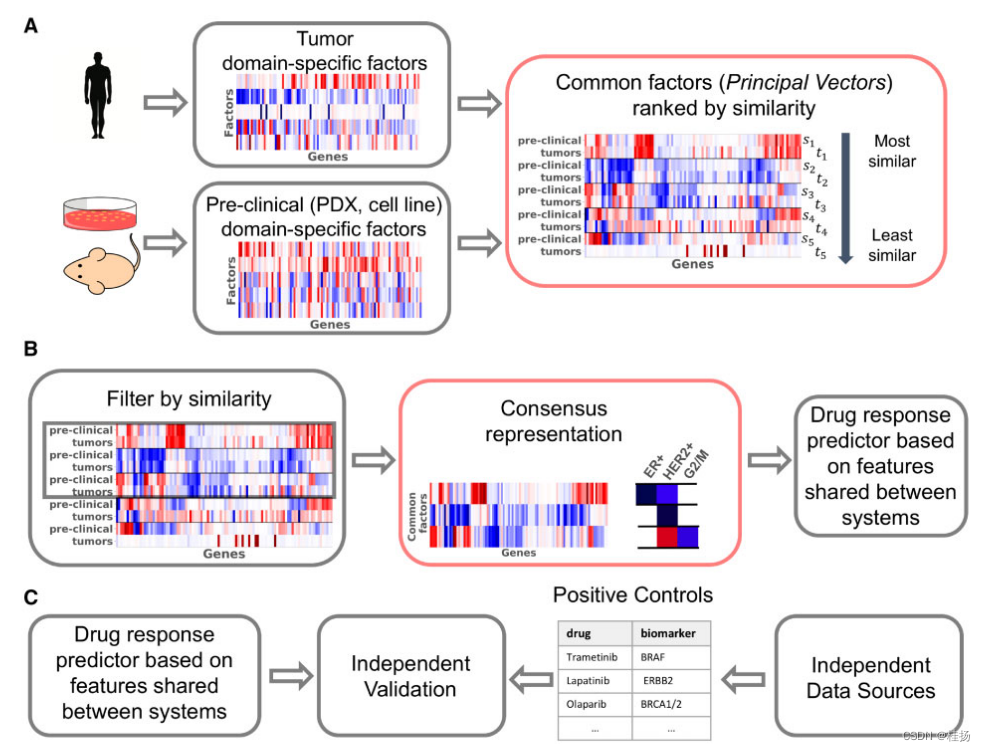
Fig. 1. Overview of PRECISE and its validation. (A)人类肿瘤和临床前数据首先被独立处理,以找到最重要的领域特定的因素(例如PCA). 然后对这些因素进行比较、排列,并按相似性排序,产生PVs. 第一个PV是一对在几何上非常相似的向量,捕捉人类肿瘤和pre- clinical模型之间的强烈共性,底部的PV代表人类肿瘤和pre- clinical模型之间的不相似性. (B) 相似性的cut-off得以保留. 对这些最相似的pre- clinical模型和肿瘤模型之间进行插值后,通过平衡人类肿瘤和pre- clinical模型,计算出一个共识表示. 我们对这些特征进行了基因集富集分析,以评估它们在临床上的相关性. 通过将pre- clinical和人类肿瘤转录组学数据投射到这个共识表示上,最终训练出一个肿瘤感知回归模型. ? 为了验证我们的模型,我们使用来自独立数据源的阳性对照,如拷贝数或突变数据。这些阳性对照是已经建立的生物标志物-药物关联。我们将我们模型的预测与基于这些独立的既定生物标志物的预测进行比较. 红框强调了我们的贡献.
这项工作包含以下新的贡献. 首先,我们引入了一种可扩展的、灵活的方法来寻找pre- clinical模型和人类肿瘤之间的共同因素. 第二,我们用这种方法来量化细胞系、PDXs和人类肿瘤之间生物过程的转录共性,并且我们表明这些共性因素在生物学上是相关的. 第三,我们展示了这些共同因素如何在回归中用于预测人类肿瘤的药物反应,并且我们恢复了著名的生物标志物-药物关联. 最后,我们推导出一种等价的、更快、更易解释的方法来计算geodesic flow kernel,这是计算机视觉中广泛使用的领域适应方法. 我们的方法建立在Gopalan等人(2011年)和Gong等人(2012年)的工作基础上,但对这些方法进行了扩展,首先是自动去除不相关的不可转移的信息,其次是在插值方案中寻找共识特征,以抵消岭回归引起的对源特征的偏见.
Materials and methods
Notes on transcriptomics data
We here present the datasets employed in this study. Further notes on preprocessing can be found in Supplementary Subsection 1.3.
The cosine similarity matrix
转录组学数据是高维的,有p ~ 19000个特征(基因),由于这些基因是高度相关的,只有一些基因的组合是有信息的. 找到这些组合的一个简单而稳健的方法(Van Der Maaten等,2009)是使用线性降维方法,如主成分分析(PCA),将数据矩阵分解为
d
f
d_f
df?个因素,分别用于源(cell lines or PDXs)和目标(human tumors),从而:
?
i
∈
{
s
,
t
}
,
X
i
=
S
i
P
i
w
i
t
h
P
i
P
i
T
=
I
d
f
(1)
\forall i \in \{ s,t \}, \qquad X_i=S_iP_i \quad with \quad P_i P_i^T = I_{d_f} \tag{1}
?i∈{s,t},Xi?=Si?Pi?withPi?PiT?=Idf??(1)
其中
s
s
s和
t
t
t分别指源和目标,
X
X
X代表(
n
×
p
n \times p
n×p)转录组学数据集,每一行代表一个样本,每一列代表一个基因.
I
d
f
I_{d_f}
Idf??是identity matrix,
P
∈
R
d
f
×
p
P \in \Bbb{R}^{d_f \times p}
P∈Rdf?×p包含行中的因子(即主成分). 由于这些因子是针对源和目标独立计算的,我们把它们称为特定领域的因子. 在这里,我们只考虑PCA,因为它被广泛采用,而且它与方差有直接联系,在分布比较中起到一阶近似的作用. 然而,我们的方法是灵活的,任何线性降维方法都可以使用.
一旦独立计算了源和目标的特定领域因子,将源因子映射到目标因子的一个简单方法是使用Fernando等人(2013)建议的subspace alignment. 这种方法找到了源因子的线性组合(
M
?
M^*
M?),尽可能地重建了目标因子:
M
?
=
arg
?
min
?
M
∈
R
d
f
×
d
f
??
∥
P
s
T
M
?
P
t
T
∥
F
=
P
s
P
t
T
(2)
M^* = \underset{M \in \Bbb{R}^{d_f \times d_f}}{{\arg\min}} \; \lVert P_s^T M - P_t^T \rVert_F = P_sP_t^T \tag{2}
M?=M∈Rdf?×df?argmin?∥PsT?M?PtT?∥F?=Ps?PtT?(2)
公式(2)是公式(1)中正交性约束下的最小二乘解. 这个最佳转换由源因子和目标因子之间的内积组成,因此量化了因子之间的相似性. 因此,我们将其称为余弦相似度矩阵(cosine similarity matrix). 它在文献中也被称为Bregman matrix divergence.
Common signal extraction by transformation analysis
正如我们将在第3.1节中显示的那样,矩阵 M ? M^* M?不是diagonal,表明源特定因素和目标特定因素之间并不存在一对一的对应关系. Moreover, using M ? M^* M? to map the source-projected data onto the target domain-specific factors would only remove source-specific variation, leaving target-specific factors and the associated variation untouched.
To understand this transformation further, we performed a singular value decomposition (SVD), i.e. P s P t T = U Γ V T P_sP_t^T=U \Gamma V^T Ps?PtT?=UΓVT, where U U U and V V V are orthogonal of size d f d_f df? and Γ \Gamma Γ is a diagonal matrix. U U U and V V V define orthogonal transformations on the source and target domain- specific factors, respectively, and create a new basis for the source and target domain-specific factors:

s 1 , … , s d f s_1, \dots ,s_{d_f} s1?,…,sdf??定义的span与source-specific factors相同, t 1 , … , t d f t_1, \dots ,t_{d_f} t1?,…,tdf??与target-specific factors也相同. 因此,PVs保留了与source-specific factors相同的信息,同时,它们的余弦相似性矩阵( Γ \Gamma Γ)是diagonal. PVs { ( s 1 , t 1 ) , … , ( s d f , t d f ) } \{(s_1,t_1), \dots ,(s_{d_f},t_{d_f})\} {(s1?,t1?),…,(sdf??,tdf??)}来自源和目标领域特定因子,并且根据它们的相似性以递减的顺序排序.
top PVs在源和目标之间非常相似,而bottom的一对则非常不相似. 由于这个原因,我们将分析限制在顶部的
d
p
v
d_{pv}
dpv? PVs. 在公式(4)中,PV被定义为使内积最大化的unitary vectors. 因此,相似度在0和1之间,可以被解释为f principal angles的余弦,定义为:
?
??
k
∈
{
1
,
?
?
,
d
f
}
,
θ
k
=
arccos
?
(
s
k
T
t
k
)
.
(5)
\forall \; k \in \{ 1, \cdots, d_f \}, \quad \theta_k = \arccos(s_k^T t_k). \tag{5}
?k∈{1,?,df?},θk?=arccos(skT?tk?).(5)
我们将
Q
s
Q_s
Qs?和
Q
t
Q_t
Qt?定义为矩阵,分别为源和目标的有序PV,其因子在行中.
Factor-level gene set enrichment analysis
为了将PV和共识表示与生物过程联系起来,我们使用了基因组富集分析(Subramanian等人,2005). 对于每个因子(即一个PV或一个共识因子),我们将肿瘤数据映射到它上面,每个肿瘤样本产生一个分数。然后在GSEA软件包中使用这些样本的分数作为连续表型. 我们采用样本级的每一个突变来评估基于1000次排列的重要性. 我们使用了MSigDB软件包中的两个curated gene sets:the canonical pathways和the chemical and genetic perturbations.
Building a robust regression model
考虑到共同因素,我们可以根据这几对PV创建一个药物反应预测器. 有不同的方法来使用这对PV. 我们可以限制在来源或目标PV上,但它只支持两个领域中的一个. 或者,我们可以同时使用源和目标PV. 然而,由于以下原因,这也将是次优的. 源PVs是使用源数据计算的,并使源的解释方差最大化. 因为目标PV没有针对源数据进行优化,投影在源PV上的源数据可能比投射在目标PV上的源数据具有更高的方差. 如果我们对投射在源和目标PV上的源数据进行惩罚性回归,它将优先选择源PV. 这反过来又会导致通用性的损失,因为源数据的信息比目标数据的信息更重要.
规避这一问题的方法之一是利用基于源和目标PV所span的空间的插值的 "中间 "特征来构建一个新的特征空间. 例如,在连接s1和t1的平面上,从前者到后者的矢量的旋转可以包含一个更好的表示. 中间特征预计是领域不变的,因为它们代表了源域和目标域之间的权衡,因此可以在回归模型中使用.
对于连接源和目标PV的中间特征,有无限多的参数化. 正如Gopalan等人(2011)和Gong等人(2012)所建议的,我们考虑the geodesic flow representing the shortest path on the Grassmannian manifold. 我们推导出(完整的证明见补充小节2.2)the geodesic的参数化,作为PV的一个函数. 定义 Π \Pi Π和 Ξ \Xi Ξ:

对于每一对PVs来说,这条geodesic path包含了在源和目标PVs之间形成rotating arc的特征. geodesic flow的优点是基于PVs,而不是特定领域的因素. 非相似的PV可以在插值之前被移除.
我们在补充小节2.3和2.4中表明,即使在无限的情况下,对所有这些特征进行投影也相当于对源和目标PV进行投影,会产生上述的不良后果. 因此,最好是为每对PV创建一个单内插特征(single interpolated feature),在源空间和目标空间所包含的信息之间取得适当的平衡. 因此,在源数据上训练的回归模型投射在内插特征上,将更好地概括目标空间. 为了构建这些内插特征或共识特征,我们使用Kolmogorov-Smirnov(KS)统计量来衡量源数据和目标数据之间的相似性,两者都投影在一个候选内插特征上.

这个过程对每一个top d p v d_{pv} dpv? PVs都要重复进行,resulting in an optimal interpolation position for each. 然后将这些位置插回geodesic curve,得到domain-invariant feature representation F F F,定义为:

Notes on implementation
Once the number of principal components and PVs have been set (see Supplementary Subsection 5 for an example), the only hyper-parameter that needs to be optimized is the shrinkage coefficient (k) in the regression model. We employed a nested 10-fold cross-validation for this purpose. Specifically, for each of the outer cross-validation folds, we employed an inner 10-fold cross-validation on 90% of the data (the outer training fold) to estimate the optimal k.To this end, in each of the 10 inner folds, we estimated the common subspace, projected the inner training and test fold on the subspace, trained a predictor on the projected inner training fold and deter- mined the performance on the projected inner test fold as a function of k. After completing these steps for all 10 inner folds, we determined the optimal k across these results. Then we trained a model with the optimal k on the outer training fold and applied the predictor to the remaining 10% of the data (outer test fold). We then employed the Pearson correlation between the predicted and actual values on the outer test folds as a metric of predictive performance. Note that every sample in an outer test folds is never employed to perform either domain adaptation nor in constructing the response predictor in that same fold.
The methodology presented in this section is available as a Python 3.7 package available on our GitHub page. The domain adaptation step has been fully coded by ourselves and the regression and cross-validation uses scikit-learn 0.19.2 (Pedregosa et al., 2011).

