现代药物发现的一个主要部分就是提出新的化合物。这项工作大多每半年进行一次,并在化学家的建议下对核心内容进行修改,通常化学家提出的这些分子中的一些子集实际上是合成并经过测试的,这个过程不断重复,直到发现合适的分子。
这个过程是很有人为局限性的。世界上没有那么多有才华的和有经验的高级化学家,所以这个过程不能向外扩展。此外根据文献报道,目前人类已知的化学空间有10^60之多,而优秀的化学家脑海中所熟悉的化学空间也是有限的,如何提高这个过程的效率,是AI制药主要解决的问题,即在庞大的化学空间中,高效准确的找到能与蛋白质靶点作用的合适化合物。
分子生成模型在提出新化合物中得到了很好的应用,目前比较常用的生成模型有两种VAE(变分自编码器)及GAN(生成对抗网络)
VAE:自编码器是试图使其输出等于输入的模型。在一个样本库中训练该模型,并调整模型参数,以便使每个样本上的输出尽可能接近输入。自编码器为瓶颈模型,如下图所示,中间层code,即瓶颈层,被称为自动编码器的潜在空间。它是样本的压缩表示空间。自动编码器的前半部分为编码器,它的工作是采集样本并将其转换为压缩表示;后半部分称为解码器,它在潜空间中获取压缩表示并将其转换会原始样本。

传统的自编码器中解码器只学会对编码器产生的特定潜在向量有效,而不是对任意向量都有效。为了改善这个问题,变分自编码器(VAE)在损失函数中添加了一项,以迫使潜在向量服从制定的分布(很多博客介绍是先验,实际上是后验)。大多数情况下,它们服从均值为0,方差为1的高斯分布。我们不会让编码器自由生成它想要的向量,而是强迫它生成一个已知分布的向量。这样,如果从相同的分布中选取随机向量,就可以期望解码器能很好的处理这些向量。
为防止过拟合,在潜在向量上添加随机噪声。
如果想详细了解VAE原理及数学知识请看这篇,知乎苏神写的VAE:
GAN:生成对抗网络(GAN)与VAE有很多共同之处,它使用相同的解码器网络将潜在向量转换为样本(只不过在GAN中,它被称为生成器而不是解码器),但以一种不同的方式训练这个网络。生成对抗网络的工作原理是将随机向量传递到生成器中,并直接评估输出是否符合预期分布,实际上,你可以创建一个损失函数来度量生成的样本与训练样本的匹配程度,然后使用该损失函数来优化模型(其实自己无法人为提出损失函数,太复杂了,实际模型是从数据中学习损失函数)。
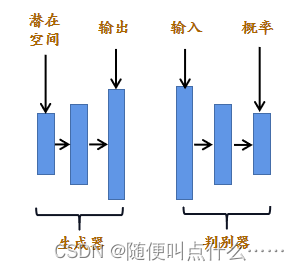
GAN由两部分组成,生成器获取随机向量并生成合成样本;第二部分称为判别器,试图将生成的样本与真实的训练样本区分开来,它以一个样本作为输入,并输出该样本是一个真实训练样本的概率。这里判别器充当了生成器的损失函数(GAN的第一篇论文2014:https://arxiv.org/pdf/1406.2661.pdf)。
两部分是同时训练的。随机向量被输入生成器,而生成器的输出被输入判别器。调整生成器的参数使判别器是输出尽可能接近1,而调整判别器的参数使其输出尽可能接近0。此外,训练集的真实样本被输入判别器,并调整其参数,使其输出接近1。
这就是“对抗”的一面。你可以把它看作是生成器和判别器之间的竞争。判别器一直在努力提高判别真假样品的能力,生成器则不断地试图在愚弄判别器方面做得更好。
VAE与GAN的区别:这两个模型各有优缺,粗略的说,GAN倾向于产生更高质量的样本,而VAE倾向于产生更高质量的分布。也就是说,由GAN生成的单个样本将更接近于训练样本,而VAE生成的样本范围更接近于训练样本范围。
重点来了,今天这篇博客,小编以2018年的一篇paper为例,介绍一下VAE在药物小分子生成中的应用:
Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
这篇paper是VAE应用在小分子生成中的开山之作,理解透彻,可以为学习分子生成模型打下基础。
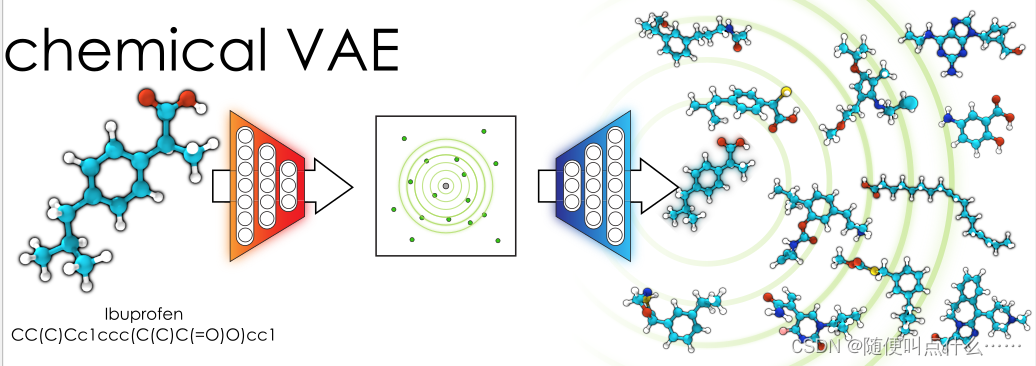
这篇文章中作者通过VAE实现了将一个离散分子表示转换成多维连续表示的方法,以用来在开放式化合物空间中产生新分子。
文章中介绍的model由三个耦合功能构建而成:
1.编码器:将分子的离散表示(smiles编码:一种分子的字符串表达,参考RDkit:介绍smiles编码,smart编码及摩根指纹(ECFP)_随便叫点什么……的博客-CSDN博客_rdkit smiles)转换为实型连续向量表示;
2.解码器:将连续向量表示转换回分子的离散表示;
3.预测器:从分子的潜在连续向量表示中估计化学性质。
背景:
虚拟筛选早已应用在药物发现中,并由此得到了许多有效的小分子药物,当前的虚拟筛选策略主要有两种:
1.搜索固定库的方法
2.离散的局部搜索法(遗传方法或离散插值方法)
通过这两种方法在已知的化学空间上搜索合适的小分子。这两种方法有一个共同的局限,就是离散数据难以有效地搜索大面积的化学空间。
目前已知的类药小分子化学空间在10^23到10^60之间(具有空间大,离散和非结构的特点为),很显然这为小分子搜索带来了难度,通过传统的虚拟筛选方式搜索小分子,已经不能满足现在的研发需求。
因此,作者提出了一种采用连续分子表示,数据驱动的方式表示复杂庞大的化学空间,并通过VAE模型实现大空间中目标分子的搜索,生成。
采用连续分子表达,数据驱动的优势:
1.首先,可以通过修改向量表示然后解码来自动生成新的化合物;
2.其次,如果开发一个从分子表示映射到所需属性的可微模型,就可以使用基于梯度的优化来在化学空间中进行更大的跳跃,基于梯度的优化可以与贝叶斯推理方法相结合,以选择可能提供有关全局最优信息的化合物;
3.最终,数据驱动的方式可以利用大量未标记的化合物自动构建更大的隐式库,然后使用较小的一组标记示例来构建从连续表示到所需属性的回归模型。这使我们能够利用包含数百万个分子的大型化学数据库,即使大多数化合物的许多特性都是未知的。
整体框架:
用生成模型来实现化学分子设计,训练一对儿深度神经网络(一个学习方差δ,一个学习均值μ)将分子(smiles编码字符串表示的分子)转化成连续的向量表示。(理论上,这种从分子表示到连续向量的转化方法同样可用于其它编码,chemical fingerprints,convolutional neural networks on graphs,similar graph-convolutions and Coulomb matrics均可)
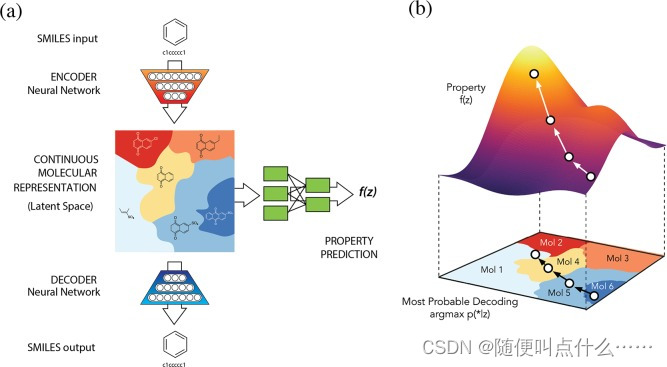
?在连续向量表示的基础上,持续优化模型来产生新颖的分子。基于属性预测任务联合训练自动编码器:添加一个多层感知器,可以预测属性值,并且把回归误差包含在损失函数中,具体过程如上面示意图所示。
训练自编码器:
自动编码器分为把字符串转换为固定长度向量的“编码器”和执行逆过程的“解码器”。对自动编码器进行训练,以使再现原始字符串时的误差最小化。自动解码器的关键在于用信息瓶颈(在这里也就是固定长度向量)来映射字符串,引导网络学习捕获数据中最显著的统计信息的压缩表示(这种表示方法捕获了数据中最显著的信息)。作者称矢量编码的分子即为该分子的潜在表示。为了使潜在空间中的无约束优化起作用,潜在空间中的点必须解码为有效的SMILES字符串,以捕捉训练数据的化学本质。否则 ,自动编码器学习得到的潜在空间就会很稀疏并且包含巨大的“死区”――即解码得到的SMILES字符串无效。为了确保潜在空间的点都能解码有效,使用变分自动解码器(VAE)框架。VAEs是作为潜变量模型的一种原则性近似推断方法发展起来的,在潜变量模型中,每个数据都有一个对应的但未知的潜在表示(这部分实际是由高斯函数和两个神经网络来实现的)。VAEs推广了自动编码器,为编码器增加了随机性(通过让所有的正态分布都像标准正态分布看齐的方式在这部分引入一定噪声,保证模型的生成能力。具体实施方法为在重构误差中加入额外的loss,加入分别代表均值?μk?和方差的对数 logσ^2的loss,当二者接近于零的时候,既接近标准正太分布,为了平衡两部分损失函数,这里加入KL散度来实现。),与惩罚项相结合,鼓励潜在空间的所有区域与有效解码相对应。当分子从潜在表示转换为分子时,解码器模型从其最后一层生成的每个位置的字符的概率分布中采样字符串。因此,单个潜在空间表示中可以有多个smiles字符串。为了实现分子设计,在自动编码器的连续表示中编码的化学结构需要与作者正在寻求优化的目标属性相关联。因此,作者在自动编码器中添加了一个多层感知器(MLP)被用来从编码分子的潜在向量中预测属性。然后,该自动编码器在重构任务和属性预测任务上进行联合训练。为了生成有希望的新候选分子,作者可以从编码分子的潜在载体开始,然后朝着最有可能改善所需属性的方向移动。由此产生新候选载体然后可以被解码成相应的分子。
(注意:使用VAE模型生成的smiles编码在化学上可能一半都是无效的,在算法模型中只是学习到了分子相似性及化学性质相似性,并没有学习或加入smiles编码的规则,因此对于模型生成的分子,还要进行进一步的筛选,除去那些不符合smiles编码及化学规则的小分子,这部分请参考我的(RDkit)其它博文。)

Figure 2?
Representations of the sampling results from the variational autoencoder. (a) Kernel Density Estimation (KDE) of each latent dimension of the autoencoder, i.e., the distribution of encoded molecules along each dimension of our latent space representation; (b) histogram of sampled molecules for a single point in the latent space; the distances of the molecules from the original query are shown by the lines corresponding to the right axis; (c) molecules sampled near the location of ibuprofen in latent space. The values below the molecules are the distance in latent space from the decoded molecule to ibuprofen; (d)?slerp?interpolation between two molecules in latent space using six steps of equal distance.
首先,分析自动编码器的保真度和潜在空间捕捉结构分子特征的能力,图2a显示了对训练集以外的5000个随机选择的ZINC分子进行编码时每个维度的核密度估计。核密度估计表示数据点沿潜在空间的每一维的分布。虽然数据点在每个单独维度上的分布显示出略有不同的平均值和标准差,但可通过变分正则化使所有的分布都是正态分布。在这个过程中也引入了一部分噪音,避免输入和输出一致。
变分自动编码器是一个双概率模型。除了添加到编码器的高斯噪声之外,因为字符串输出是从解码器的最后一层采样的,故解码过程也是不确定的。这意味着解码潜在空间中的单个点返回到字符串表示是随机的。图2b显示了将FDA批准的样本药物分子的潜在表示解码为几种不同分子的可能性。对于大多数潜在点,一个最有可能的分子将被解码,而其他许多细微的变化则以较低的频率出现。当将这些生成的SMILES重新编码到潜在空间中时,解码次数最多的也往往是到原始点的欧式距离最小的点,这表明潜在空间的确捕获了与分子相关的特征。
图2c显示了潜伏空间中一些接近布洛芬的分子。随着潜在空间中距离的增加,这些结构与布洛芬变得不那么相似。当距离接近训练集中分子的平均距离时,变化会更加明显,最终类似于可能从训练集中抽样的随机分子。从潜在空间中的一个点解码的概率取决于该点与其他分子的潜在表示的距离;作者观察到,对于接近已知分子的点,解码率为73?79%,对于随机选择的潜伏点,解码率为4%。
连续的潜在空间允许分子按照其潜在表示之间的最短欧几里得路径进行内插。在探索高维空间时,重要的是要注意,欧几里得距离可能不会直接映射到分子的相似性概念。在高维空间中,大多数独立的正态分布随机变量的质量不在平均值附近,而是在平均值周围的环形空间中。两点之间的线性插值可能会经过一个低概率的区域,为了保持对高概率区域的采样,作者使用了球面内插(Slerp)。使用slerp时,两点之间的路径是位于N维球体表面上的圆弧。图2d显示了两个随机药物分子之间的球面内插,显示了其间的平滑过渡。
分子的性质预测:

?Figure 3?
Two-dimensional PCA analysis of latent space for variational autoencoder. The two axis are the principle components selected from the PCA analysis; the color bar shows the value of the selected property. The first column shows the representation of all molecules from the listed data set using autoencoders trained without joint property prediction. The second column shows the representation of molecules using an autoencoder trained with joint property prediction. The third column shows a representation of random points in the latent space of the autoencoder trained with joint property prediction; the property values predicted for these points are predicted using the property predictor network. The first three rows show the results of training on molecules from the ZINC data set for the logP, QED, and SAS properties; the last two rows show the results of training on the QM9 data set for the LUMO energy and the electronic spatial extent (R2).
人们对发现新分子的兴趣通常与最大化某些理想的性质有关。为此,作者扩展了纯生成模型,使其也能从潜在表示中预测属性值。作者训练了一个多层感知器和自动编码器,以根据每个分子的潜在表示来预测性质。通过性质预测的联合训练,分子在潜在空间中的分布按属性值组织。图3显示了使用PCA将属性值映射到分子的潜在空间。由自动编码器与属性预测任务共同训练而产生的潜在空间在分子分布中通过属性值显示了一个梯度。具有高值的分子位于一个区域中,而具有低值的分子位于另一个区域中。在没有属性预测任务的情况下接受训练的自动编码器不会在最终的潜在表示分布中显示出关于属性值的可识别模式。
在VAE模型中可调节的参数:
1.对循环/卷积编码器的选择
2.隐藏层数
3.层的大小
4.正则化
5.学习率
代码部分请参考:
https://github.com/aspuru-guzik-group/chemical_vae![]() https://github.com/aspuru-guzik-group/chemical_vae补充:
https://github.com/aspuru-guzik-group/chemical_vae补充:
此处介绍两个参数QED及SA。
QED:定量估计药物相似度参数(QED),QED最早是由Bickerton提出的(引文1),通过将计算出的每个分子与一组性质与上市药物中相同性质的分布比较来给予打分。这个分数在0~1之间,值接近1的被认为更接近药物。
SA:合成可及性得分(SA).范围为0-10,数值越小,说明越容易合成。
获得一个分子的smiles编码后,通过rdkit可计算出QED及SA,一般进行筛选时,QED>=0.6,SA<=4的范围比较合适,这个范围仅供参考,根据具体情况设定 。
每周一更,总结不易,如果对您有帮助,希望能点个赞,多谢。
引文: