�����ע,�������
��������ں��Ƴ���ϸ��ϵ�н̳�,����Ҫ���ŷ�������ʦ������ϵ����!��ѡ����ת¼�����̳���������:
SCS��1�����쿪����ϸ��֮��,��˵��ϸ�������ǰ������
SCS��2����ϸ��ת¼�� ֮ cellranger
SCS��3����ϸ��ת¼������ GEO ���ؼ���ȡ
SCS��4����ϸ��ת¼�����ݿ��ӻ����� (Seurat 4.0)
SCS��5����ϸ��ת¼�����ݿ��ӻ����� (scater)
SCS��6����ϸ��ת¼��֮ϸ�������Զ�ע�� (SingleR)
SCS��7����ϸ��ת¼��֮�켣���� (Monocle 3) ���ࡢ����ͼ���ϸ��
������˵˵��ϸ��ת¼�����ݵ�ϸ���켣����,ѧ����Щ�������,���뷢���¾�ֻ��������ѡ����,�д����Ե���������Ϊ���µ�����,�����Ƿ���������!
ǰ ��
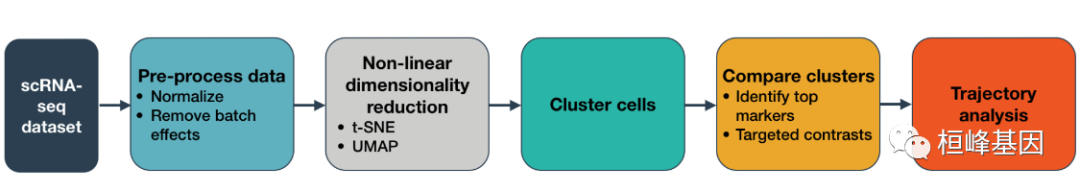
��ϸ��ת¼�����(scRNA-seq)ʵ��ʹ�����ܹ������µ�ϸ������,�����������˽�����������ڷ��������в����ġ�Monocle 3���ṩ��һ��������ϸ���������ʵ��Ĺ��߰���
Monocle 3����ִ��������Ҫ���͵ķ���:
-
���ࡢ����ͼ���ϸ������ϸ��RNA-Seqʵ�����������µ�(�����Ǻ�����)ϸ�����͡�
-
������ϸ���켣���ڷ�������������������������,ϸ����һ��״̬���ɵ���һ��״̬��Monocle 3���Է�����Щת�䡣
-
����������������ϸ�����ͺ�״̬������,����Ҫ�����������������ϸ�����бȽϡ�Monocle 3����һ�����ӵ�,������ʹ�õı���ϵͳ��
Monocle 3����Ҫ����
Monocle 3�ѱ��������,���ڷ������͡����ӵĵ�ϸ�����ݼ���Monocle 3�ĺ����㷨���и߶ȵĿ���չ��,���Դ����������ϸ����Monocle 3������һЩǿ����¹���,ʹ���������̥��ģ��ʵ�������Ϊ����:
-
һ�����õĽṹ������������ѧϰ��չ�켣;
-
֧��UMAP�㷨��ʼ���켣�ƶ�;
-
֧�ֶ���켣;
-
ѧϰ��ѭ����������켣�ķ���;
-
�Զ��ָ�ϸ�����㷨,���á�����ͼ����˼����ѧϰ���ཻ��ƽ�еĹ켣;
-
һ���µĻ������켣������ͳ�Ʋ���;
-
����ѯ����ӳ�䵽������;
-
��ע�ʹ�����ת�Ƶ���ѯ���ݼ�;
-
���沢����Monocle�����ת��ģ��;
-
fit_models�Ļ�ϸ�����ֲ�;
-
һ�����ӻ��켣�ͻ�������3D���档
��������ͼ����:
������װ
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.14")
BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats',
'limma', 'lme4', 'S4Vectors', 'SingleCellExperiment',
'SummarizedExperiment', 'batchelor', 'Matrix.utils',
'HDF5Array', 'terra', 'ggrastr'))
install.packages("devtools")
devtools::install_github('cole-trapnell-lab/monocle3')
���ݶ�ȡ������
Monocle��cell_data_set��Ķ����б��浥ϸ������ʽ���ݡ�����������Bioconductor SingleCellExperiment��,�����ṩ��һ��ͨ�ýӿ�,������Щʹ��Bioconductor����������ϸ��ʵ�������˵�Ǻ���Ϥ�ġ��������Ҫ���������ļ�:
-
expression_matrix,����ֵ�����־���,���ǻ���,����cell
-
cell_metadata,һ�����ݿ�,����cell,����cell����(��ϸ������,��������,������);
-
gene_metadata,һ�����ݿ�,��������(�����),���ǻ�������,����������,gc���ݵȡ�
����ֵ�������:
(1). ӵ����cell_metadata��������ͬ������;
(2). ӵ����gene_metadata��������ͬ��������
����:
-
cell_metadata:�����������Ӧ�������ʽ���������ƥ��;
-
gene_metadata:���������Ӧ��ƥ�����ʽ���������;
-
gene_metadata:һ��Ӧ������Ϊ��gene_short_name��,������ÿ������Ļ�����Ż������(ͨ�����ڻ�ͼ)��
Monocle3 ����:
https://cole-trapnell-lab.github.io/monocle3/
����pbmc���Ƿֻ����������ϸ��,�����ϲ�������ֱ�ӵķֻ���ϵ,��˲��ʺ���������ʱ�켣����������ֻ��ʹ���������Դ������ݼ�����ѧϰ��ʾ��
�ٷ����Ľ̳���ֱ����ȡ,�����������ǹ��ڶ�ȡ�ٶȷdz���,�Ұ�����rds��������,����Ҫ���Ե���ʦ��,���Լ�����,˽�Ÿ���!
library(monocle3)
# Load the data expression_matrix <-
# readRDS(url('https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/cao_l2_expression.rds'))
# cell_metadata <-
# readRDS(url('https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/cao_l2_colData.rds'))
# gene_annotation <-
# readRDS(url('https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/cao_l2_rowData.rds'))
expression_matrix <- readRDS("cao_l2_expression.rds")
cell_metadata <- readRDS("cao_l2_colData.rds")
gene_annotation <- readRDS("cao_l2_rowData.rds")
Step 1: Normalize and pre-process the data
ʹ��Monocle 3�ĵ�һ���ǽ����ݼ��ص�Monocle 3������cell_data_set:
# Make the CDS object
cds <- new_cell_data_set(expression_matrix, cell_metadata = cell_metadata, gene_metadata = gene_annotation)
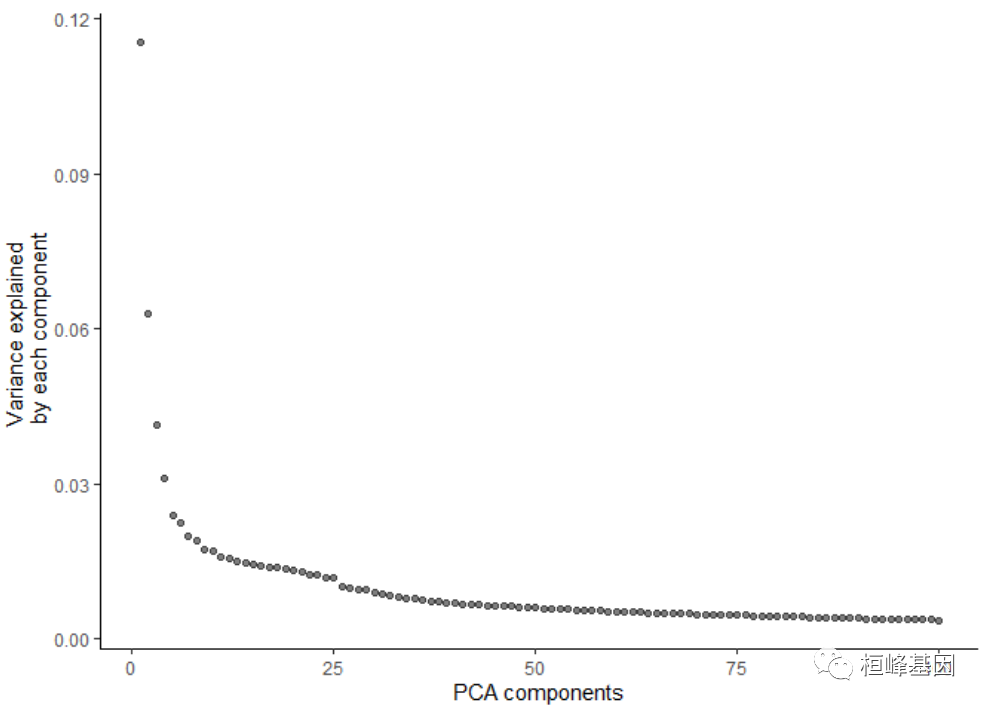
cds <- preprocess_cds(cds, num_dim = 100, method = c("PCA", "LSI"))
plot_pc_variance_explained(cds)
Step 2: Remove batch effects with cell alignment
��Monocle 3��,����ʹ�ü��ֲ�ͬ�ķ���������(��������ȫ��ͬ)�������м�ȥδ�۲쵽������ЧӦ������ϸ����
cds <- align_cds(cds, alignment_group = "batch")
Step 3: Reduce the dimensions using ��UMAP��, ��tSNE��, ��PCA��, ��LSI��, ��Aligned��
��ά�㷨,�������ṩ��5�ַ���:
cds <- reduce_dimension(cds, reduction_method = c("UMAP", "tSNE", "PCA", "LSI", "Aligned"))
Step 4: Cluster the cells
ϸ������:
cds <- cluster_cells(cds)
Setp 5: Visualization
�������ݷֲ�
��������,����ʹ��Monocle����Ҫ���ƺ���plot_cells():
plot_cells(cds)
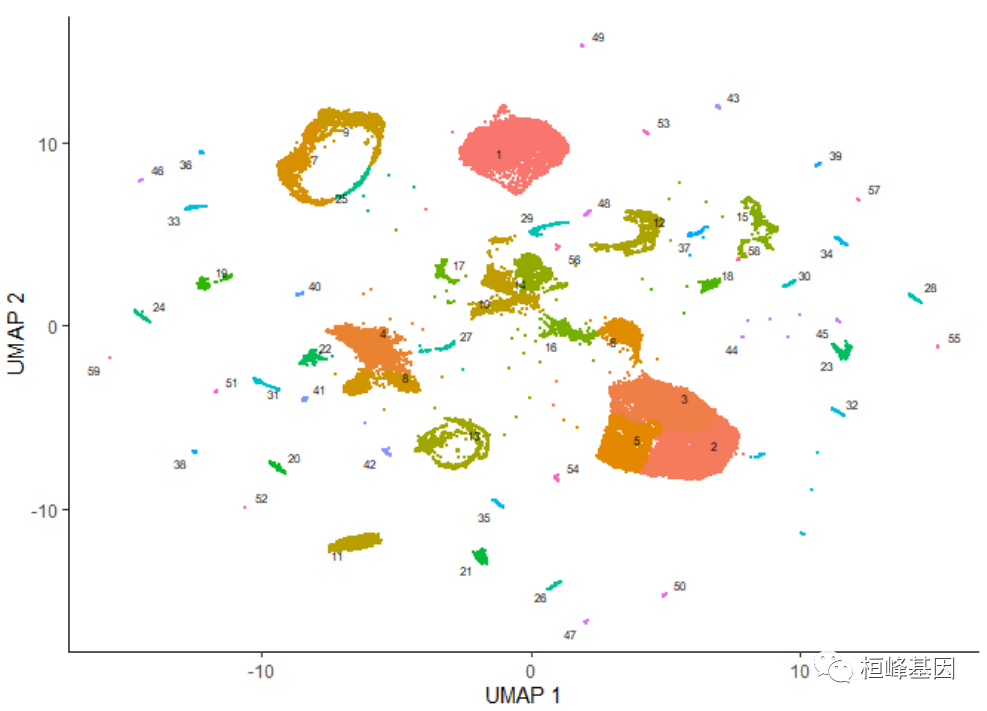
����ϸ������
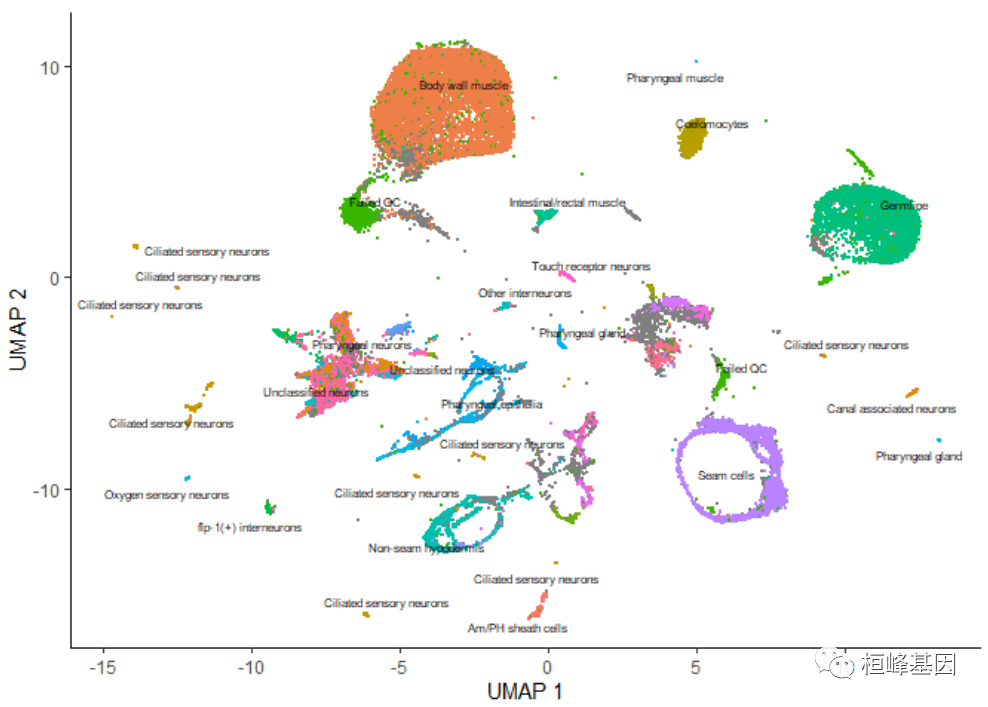
��ͼ�е�ÿ�����ʾcell_data_set����cds�е�һ����ͬ��ϸ������������������,��Щϸ�������������,��Щ����ǧ��ϸ��,��Щֻ�м�����ͨ���۲�������Ļ���,���������ֹ�ע��ÿ��ϸ�������ǿ���ʹ��plot_cells()��color_cells_by����ͨ�����ߵ�ԭʼע��UMAPͼ�еĵ�Ԫ����ɫ��
plot_cells(cds, color_cells_by = "cao_cell_type")
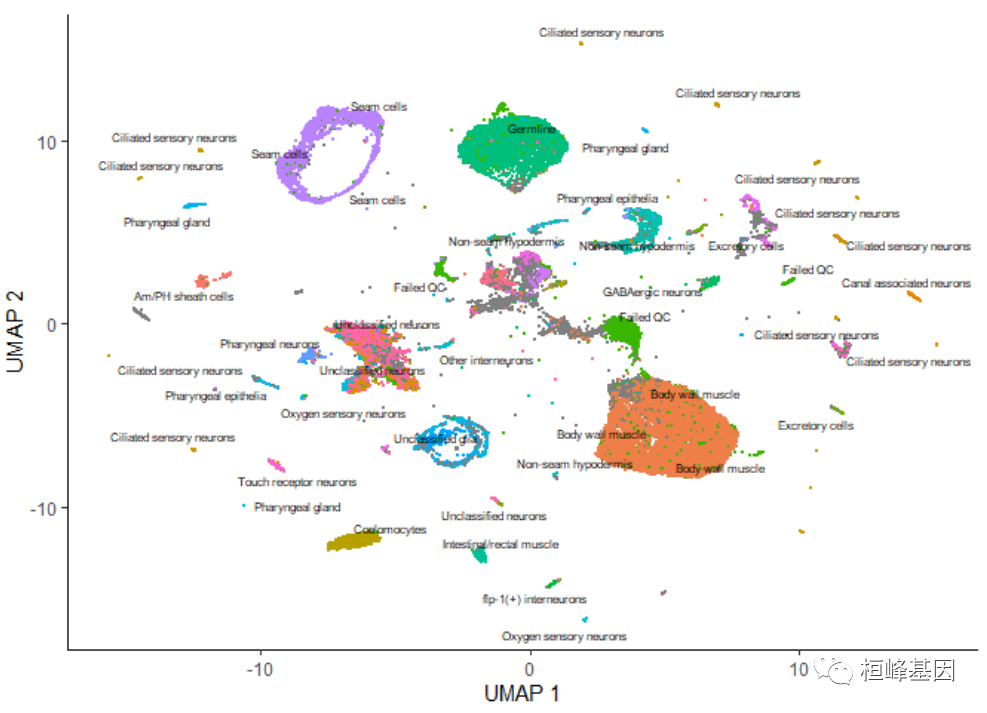
������ɫ
��UMAPͼ��,����Կ�������ϸ�����ͷdz��ӽ��������Ժ�������һЩ�����,color_cells_by������colData(cds)���κ��е����ơ�ע��,��color_cells_by��һ���������ʱ,��ǩ�������ӵ���ͼ��,ÿ����ǩ����λ�ھ��иñ�ǩ�����е�Ԫ����м䡣
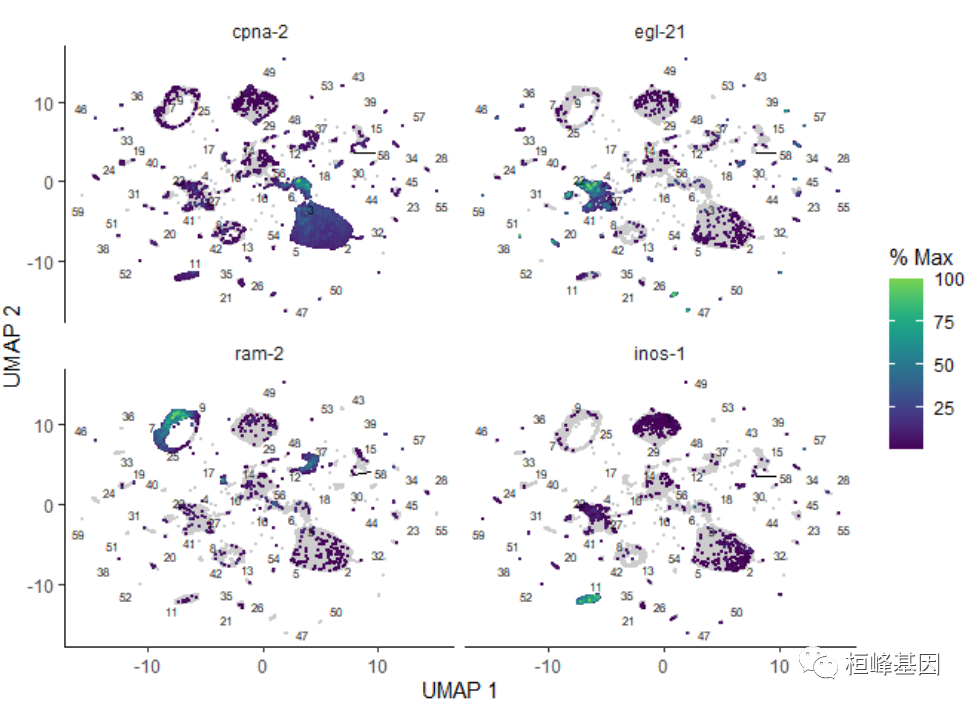
��Ҳ���Ը���ϸ������Ļ����һ�����Ķ�������ϸ����ɫ:
plot_cells(cds, genes = c("cpna-2", "egl-21", "ram-2", "inos-1"))
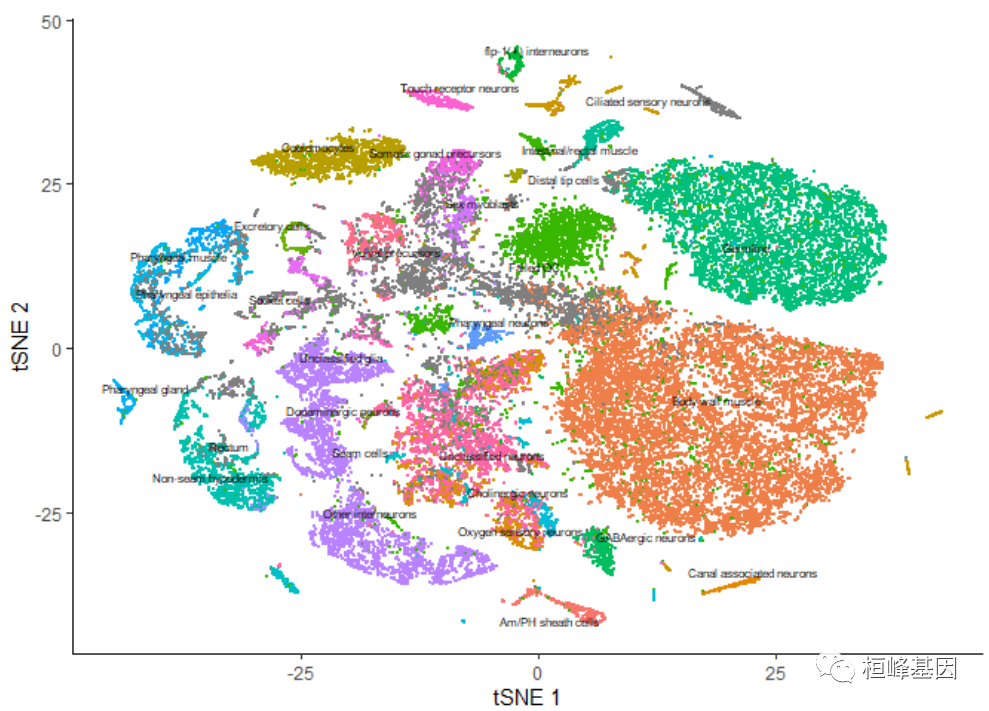
tSNE��ά��ͼ
cds <- reduce_dimension(cds, reduction_method = "tSNE")
plot_cells(cds, reduction_method = "tSNE", color_cells_by = "cao_cell_type")
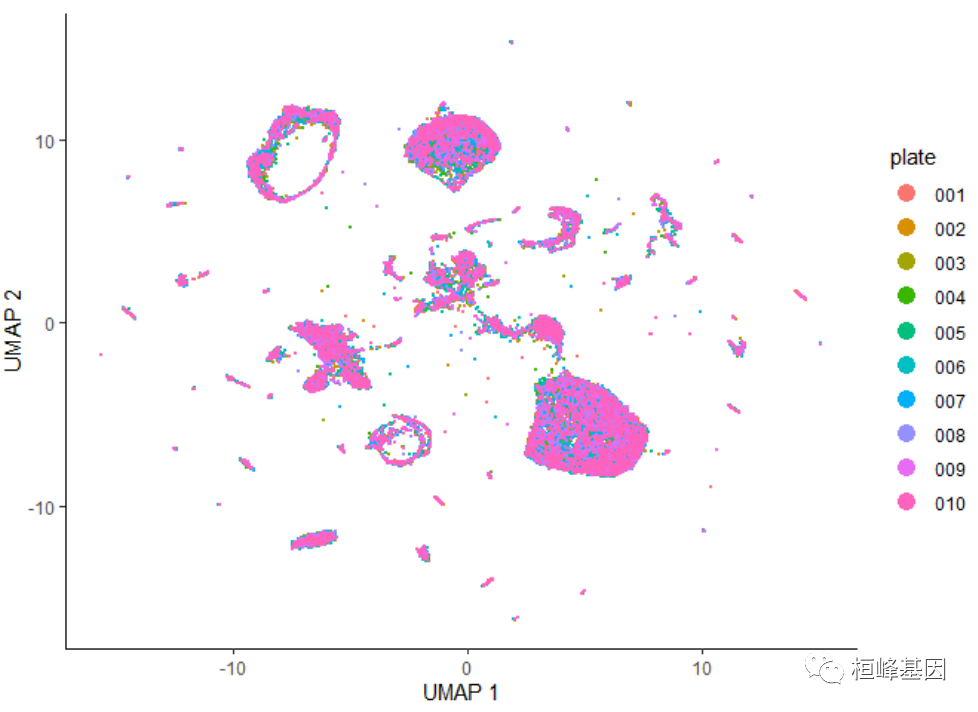
���ȥ������ЧӦ
�ڽ��л���������ʱ,����ЧӦ�Ǻ���Ҫ��,����ЧӦ��ָ��ͬʵ����������ϸ��ת¼���ϵͳ�Բ��졣��Щ�����Ǽ����Ե�,���ڵ�ϸ��RNA-seqЭ���������,������ѧ��,��������Բ�ͬ��С�����Щ�����ʶ��������Ч������������,�Ӷ�ʹ���Dz���������ķ���,����һ�����ӵ�����,��Monocle�ṩ�˴������ǵĹ��ߡ�
����ɫ����ɫ
��ִ�н�άʱ,Ӧ��ʼ�ռ��������Ч������Ӧ����colData����һ����,���ж�ÿ����Ԫ�������ĸ����������б��롣Ȼ��,�����Լ�ͨ����������ϸ����ɫ���������м�����һ������顱ע��,ָ����ÿ��ϸ�������ĸ���ѧ RNA - SEQ��顣��ɫ����ɫ UMAP ��ʾ:
plot_cells(cds, color_cells_by = "plate", label_cell_groups = FALSE)
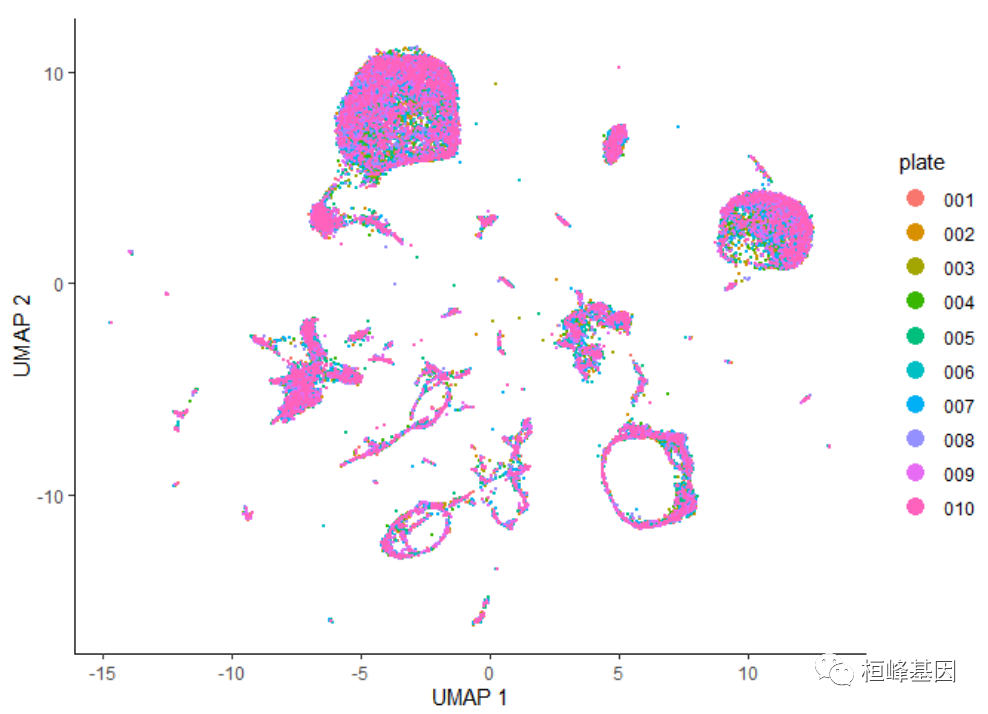
align_cds() ȥ������ЧӦ
��Щ�����в�û�����Ե�������Ч������������а������������������������ʵ���Ա仯,���Ǿͻ���������ʵ����ֻ����һ���������ϸ��Ⱥ��Ȼ��,���ǿ��Գ���ͨ������align_cds()������ɾ����������Ч��:
cds <- align_cds(cds, num_dim = 100, alignment_group = "plate")
cds <- reduce_dimension(cds)
plot_cells(cds, color_cells_by = "plate", label_cell_groups = FALSE)
��ϸ������ɴ�
��ϸ������Ϊ cluster ��ʶ�������б���ϸ�����͵���Ҫ���衣Monocleʹ��һ�ֳ�Ϊ�������ļ�������ϸ�����з��顣Levine�������������ַ���,��Ϊ����ͼ�㷨��һ���֡������ʹ��cluster_cells()����������ϸ��,��������:
cds <- cluster_cells(cds, resolution = 1e-05)
plot_cells(cds)
ע��,���ڵ����ǵ��ò���������plot_cells()ʱ,�������Ĭ��ֵ�������ϸ����ɫ��
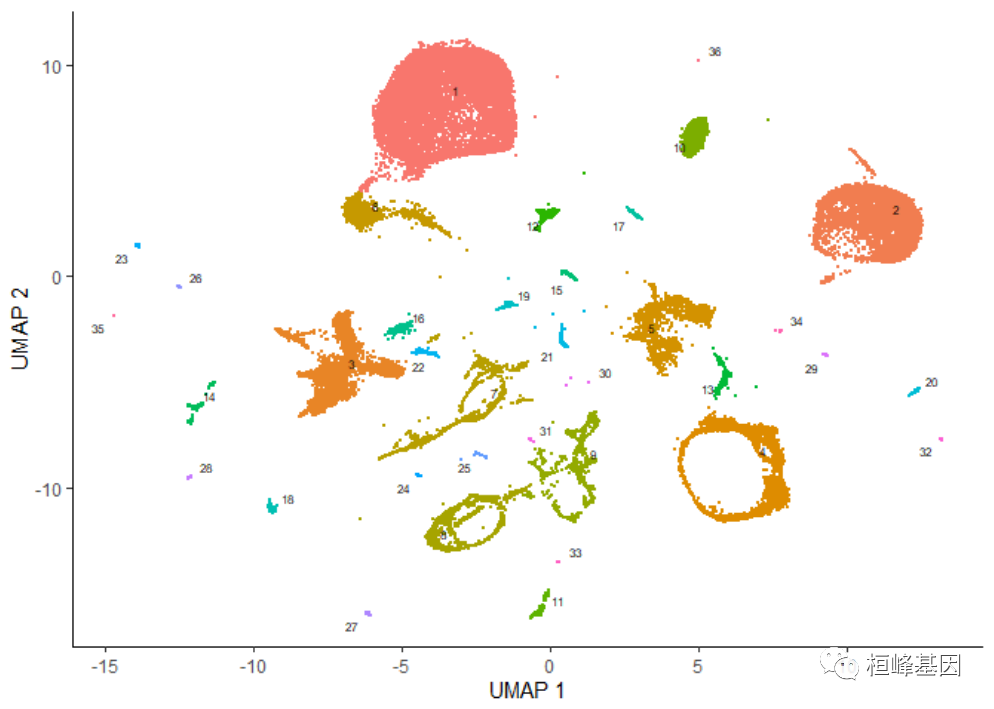
cluster_cells()��ʹ��Alex Wolf������ΪPAGA�㷨��һ���������ͳ�Ʋ���,��ϸ���ֳɸ����������,��Ϊ������������������ӻ���Щ����:
plot_cells(cds, color_cells_by = "partition", group_cells_by = "partition")
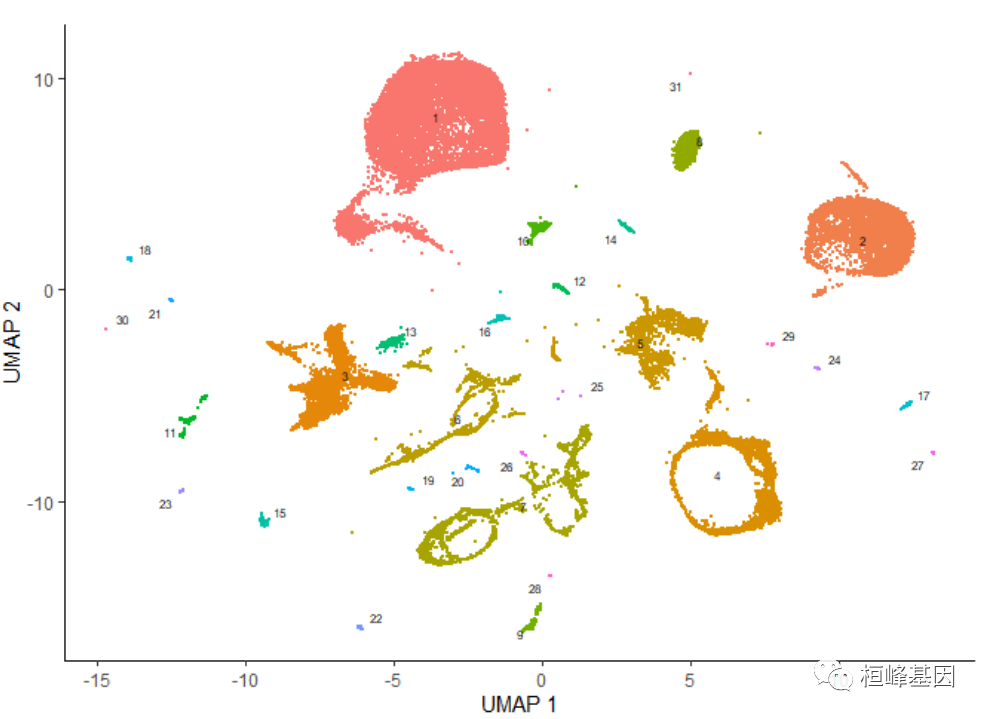
һ������cluster_cells(), plot_cells()��������������Ҫ��ϸ����ɫ�ķ�ʽ��ÿ��ϸ���ؽ��е�����ǡ�����,����ĵ��ø������ǵ�ϸ������ע�Ͷ�ϸ��������ɫ,ÿ���ظ������������ע�ͽ��б��:
plot_cells(cds, color_cells_by = "cao_cell_type")
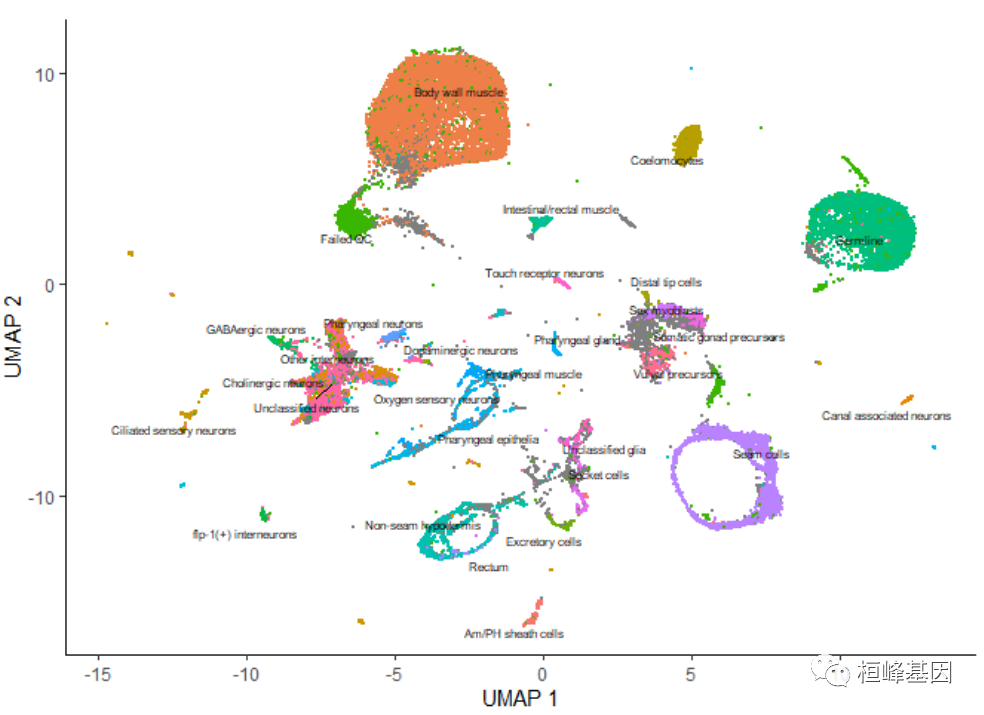
ͨ������ group_cells_by=��partition��,����ѡ�����������������Ǵء���������ͨ���� labels_per_group=2 ���ݸ� plot_cells() ������ÿ����Ⱥ��ǰ2����ǩ�����,���Խ��������Dz���,ʹ plot_cells() ����� cluster_cells() ֮ǰһ��,������ʾ:
plot_cells(cds, color_cells_by = "cao_cell_type", label_groups_by_cluster = FALSE)
���������ȷ�����һ����,���ݹ���,һ������е�̫����,Ŀǰ��ϸ������ķ���Ҳ�ڽ���,��ϸ��ϵ�п�����Ŀǰ�IJ�������,���ⷽ���������ʦ,��ϵ�������,�ṩ��߶˵Ŀ��з���!
�������,����ɹ�����!
δ����������ںŽ�����ϵ��Ƴ���ϸ��ϵ�����ŷ����̳�,
�����ڴ�!!
��������Ž���Ⱥ����ʦ����ɨ���һ����ά�����,��ע����λ+����+Ŀ�ġ�,��Щ�뷢���ľ�����Ű�,���÷����������߳�ȥ!
References:
-
UMAP: McInnes, L, Healy, J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction, ArXiv e-prints 1802.03426, 2018
-
tSNE: Laurens van der Maaten and Geoffrey Hinton. Visualizing data using t-SNE. J. Mach. Learn. Res., 9(Nov):2579�C 2605, 2008.