�����ע,�������
��������ں��Ƴ�ת¼��������ٴ�Ԥ��ģ�ͽ̳�,����Ҫ���ŵ���ʦ������ϵ����!��ѡ����ת¼�����̳���������:
RNA 1. ���������Щ�¨C���� GEO
RNA 2. SCI�����л���GEO�IJ���������֮ limma
RNA 3. SCI �����л���T CGA ����������֮ DESeq2
RNA 4. SCI �����л���TCGA �������֮ edgeR
RNA 5. SCI �����в���������֮ MA ͼ
RNA 6. ����������֮-- ��ɽͼ (volcano)
RNA 7. SCI �����еĻ����������ɷַ��� (PCA)
RNA 8. SCI�����в���������C��ͼ (heatmap)
RNA 9. SCI �����л������֮ GO ע��
RNA 10. SCI �����л�����︻��֮�CKEGG
RNA 11. SCI �����л�����︻��֮ GSEA
RNA 12. SCI �������������߽�����㷽��֮ CIBERSORT
RNA 13. SCI �����в���������֮ WGCNA
RNA 14. SCI �����в���������֮ ���������� (PPI)
RNA 15. SCI �����е��ںϻ���֮ FusionGDB2
RNA 16. SCI �����е��ںϻ���֮���ӻ�
RNA 17. SCI �����е�ɸѡ Hub ���� (Hub genes)
RNA 18. SCI ������������ GSVA
RNA 19. SCI �������ල���෨ (ConsensusClusterPlus)
RNA 20. SCI �����е��������߽������ (ssGSEA)
RNA 21. SCI �������������
RNA 22. SCI �����л��ڱ�����ƶ���������֯�Ļ���ϸ��������ϸ��(ESTIMATE)
RNA 23. SCI�����б������ģ�͵ķ������ӹ���ͼ(ggrisk)
RNA 24. SCI�����л���TCGA�����߽���ϸ������ (TIMER)
RNA 25. SCI�����й�����֯��������ϸ���ͻ���ϸ��Ⱥ��Ⱥ����(MCP-counter)
RNA 26. SCI�����л���ת¼�����ݵĻ�����������ƶ� (GENIE3)
�ٴ�Ԥ��ģ����������:
Topic 1. �ٴ���־�����ŷ�������˼·
Topic 2. �������֮ Kaplan-Meier
Topic 3. SCI���µ�һ�ű���C���߱���
Topic 4. �ٴ�Ԥ��ģ���� Logistic �ع�
Topic 5. ������ȷ�����ָ�
Topic 6 �����������ɻع�
Topic 7. �ٴ�Ԥ��ģ�ͨCCox�ع�
Topic 8. �ٴ�Ԥ��ģ��-Lasso�ع�
Topic 9. SCI ���µڶ��ű��������ػع������
Topic 10. ������ Logistic �ع�����������ط�������
Topic 11. SCI�ж�Ԫ����ɸѡ����/�����ر�
Topic 12 �ٴ�Ԥ��ģ�͡����߱� (Nomogram)
Topic 13. �ٴ�Ԥ��ģ�͡�һ����ָ�� (C-index)
Topic 14. �ٴ�Ԥ��ģ��֮У���� (Calibration curve)
Topic 15. �ٴ�Ԥ��ģ��֮�������� (DCA)
Topic 16. �ٴ�Ԥ��ģ��֮�����߲����������� (ROC)
Topic 17. �ٴ�Ԥ��ģ��֮ȱʧֵʶ���ӻ�
Topic 18. �ٴ�Ԥ��ģ��֮ȱʧֵ�岹����
�������һ�»���ת¼�����ݵĻ�����������ƶ�,��������˾ٸ�������,���о�һ����ϸ��ת¼�������,���л�����������ƶ�,Ч�����Dz�����,�������ܹ������ǵĵ�ϸ��������bulk�����ݽ������,��ȱ��©,ʹ�������ϸ�������,����˵����!!!
ǰ ��
����ϵͳ����ѧ������ü�Ŀ�������֮һ�����ø�ͨ������������(�ر��ǻ����������)�����Ŵ���������(GRNs)�����˽ṹ�� ���������ͷ����Ի�(DREAM)��սּ������GRN�����㷨��ģ�����ݻ��ϵijɹ��̶ȡ�һ���µ�GRN�����㷨GENIE3,���㷨��DREAM4 In silica��������ս�б�����á�GENIE3��p���������������Ԥ��ֽ�Ϊp����ͬ�Ļع����⡣��ÿ���ع�������,���û������ļ��ɷ���Random Forests��Extra-Trees,��������������(�������)�ı���ģʽ��Ԥ������һ������(Ŀ�����)�ı���ģʽ�����������Ԥ��л������ģʽ�е���Ҫ�Ա���Ϊ�ٶ��ĵ��ػ��ڵ�ָʾ������ĵ������ӱ��ۺϵ����л�����,�ṩһ������õ�����,�������������ؽ��������硣������DREAM4 In silica��������սģ�������б���������,������ GENIE3������˾�����������緽�����������㷨����û�жԻ�����صı������κμ���,���Դ�����Ϻͷ����Ե������,��������GRN,���ٺͿ���չ����֮,�����һ���µ�GRN�����㷨,���㷨�ںϳɺ���ʵ������������϶��������á����㷨�������ļ��ɷ���������ѡ��,��ͨ��,�������������͵Ļ��������ݺͽ�����
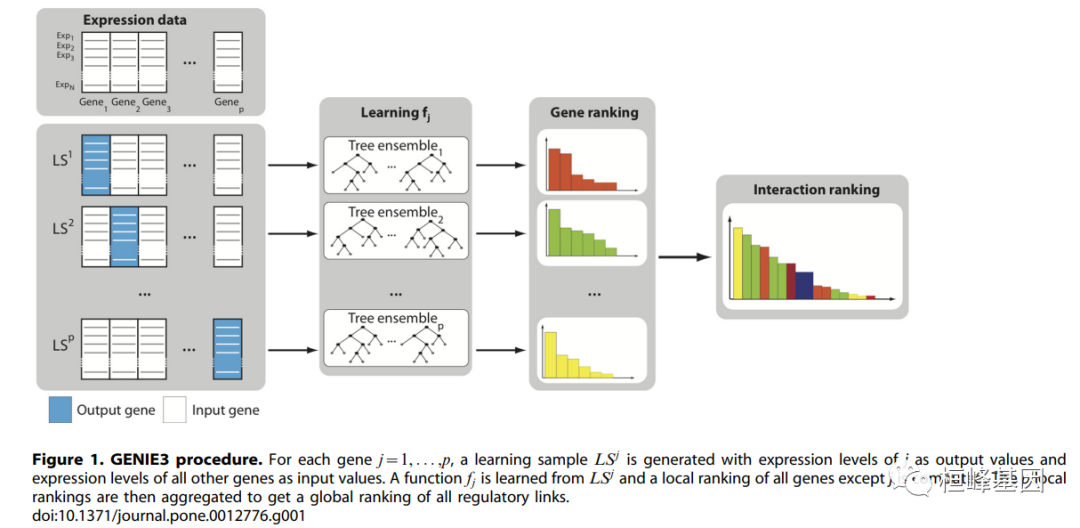
��������װ
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GENIE3")
����ʵ��
�������Dz���GENIE3������������,�������Dz��� ��ϸ��ת¼������ݼ� pbmc ���Է������ڵ�ϸ��ת¼�����ݵĻ����������Ĺ�����
GENIE3����
����һ���������ݼ�
exprMatr <- matrix(sample(1:10, 100, replace = TRUE), nrow = 20)
rownames(exprMatr) <- paste("Gene", 1:20, sep = "")
colnames(exprMatr) <- paste("Sample", 1:5, sep = "")
head(exprMatr)
## Sample1 Sample2 Sample3 Sample4 Sample5
## Gene1 4 9 5 1 8
## Gene2 6 3 10 5 5
## Gene3 2 2 3 6 7
## Gene4 5 5 2 4 7
## Gene5 1 8 2 1 1
## Gene6 8 2 7 7 3
treeMethod: Tree-based method used. Must be either ��RF�� for Random Forests (default) or ��ET�� for Extra-Trees.
������������������ʱ�����ַ�������ѡ��:RF, ET,Ĭ���� RF��
���л����������ķ���
library(GENIE3)
set.seed(123) # For reproducibility of results
weightMat <- GENIE3(exprMatr)
dim(weightMat)
## [1] 20 20
weightMat[1:5, 1:5]
## Gene1 Gene2 Gene3 Gene4 Gene5
## Gene1 0.00000000 0.044491854 0.02449400 0.06013293 0.112097046
## Gene2 0.03646480 0.000000000 0.02942083 0.07192433 0.129756000
## Gene3 0.02743010 0.008567765 0.00000000 0.05837880 0.013269916
## Gene4 0.04856294 0.121335689 0.05750308 0.00000000 0.004207474
## Gene5 0.04370138 0.032089715 0.02260877 0.01166890 0.000000000
���ú�ѡ����:
# Genes that are used as candidate regulators
regulators <- c(2, 4, 7)
# Or alternatively:
regulators <- c("Gene2", "Gene4", "Gene7")
weightMat <- GENIE3(exprMatr, regulators=regulators)
regulatorsList <- list("Gene1"=rownames(exprMatr)[1:10],
"Gene2"=rownames(exprMatr)[10:20],
"Gene20"=rownames(exprMatr)[15:20])
set.seed(123)
weightList <- GENIE3(exprMatr, nCores=1, targets=names(regulatorsList), regulators=regulatorsList, returnMatrix=FALSE)
ѡ�� ET ��������:
# Use Extra-Trees (ET) method
# 7 randomly chosen candidate regulators at each node of a tree
# 5 trees per ensemble
weightMat <- GENIE3(exprMatr, treeMethod="ET", K=7, nTrees=50)
head(weightMat)
## Gene1 Gene2 Gene3 Gene4 Gene5 Gene6
## Gene1 0.00000000 0.003047264 0.031212121 0.011919192 0.09888889 0.23666667
## Gene2 0.04398058 0.000000000 0.028939394 0.050858586 0.08549283 0.04578767
## Gene3 0.02697411 0.020870647 0.000000000 0.040580808 0.01430108 0.01815068
## Gene4 0.01642395 0.163868159 0.088939394 0.000000000 0.06030466 0.05730594
## Gene5 0.01677994 0.075410448 0.045151515 0.003787879 0.00000000 0.07142694
## Gene6 0.23944175 0.047164179 0.009545455 0.009090909 0.07493728 0.00000000
## Gene7 Gene8 Gene9 Gene10 Gene11 Gene12
## Gene1 0.018822844 0.032118644 0.007031250 0.010097324 0.054166667 0.006493506
## Gene2 0.031235431 0.076292373 0.011979167 0.014251825 0.177222222 0.024567100
## Gene3 0.060081585 0.070621469 0.007890625 0.190681265 0.006666667 0.016428571
## Gene4 0.009026807 0.029131356 0.021562500 0.116465937 0.145000000 0.012694805
## Gene5 0.007167832 0.169258475 0.105729167 0.006739659 0.013333333 0.014404762
## Gene6 0.001456876 0.002542373 0.024218750 0.000000000 0.031111111 0.007142857
## Gene13 Gene14 Gene15 Gene16 Gene17 Gene18
## Gene1 0.124664352 0.006725146 0.02627148 0.016343042 0.05244624 0.03888889
## Gene2 0.006018519 0.115730994 0.07525773 0.013122977 0.04456989 0.01861111
## Gene3 0.039479167 0.013742690 0.02482818 0.005315534 0.01247312 0.04722222
## Gene4 0.032557870 0.039605263 0.03470790 0.017467638 0.02643369 0.04000000
## Gene5 0.006944444 0.034327485 0.25359107 0.008171521 0.18548387 0.02555556
## Gene6 0.018865741 0.003070175 0.07048110 0.020161812 0.01462366 0.01944444
## Gene19 Gene20
## Gene1 0.08717172 0.0376548673
## Gene2 0.09570076 0.0121681416
## Gene3 0.15281566 0.0987020649
## Gene4 0.05696970 0.1382448378
## Gene5 0.03516414 0.0002212389
## Gene6 0.09481061 0.0234070796
��ȡtop link
����õ��������к�ѡ����Ŀ��ܵ�������,��һ���dz���ı��������Ļ���list�м��ٸ�����,���õ�ʮ�������ʮ���link,���Ѵ��л����Ч��Ϣ����ʱ�����ѡ��ֻ��ȡtop link,�����п��ܵĵ��ع�ϵ���硣
set.seed(123) # For reproducibility of results
weightMat <- GENIE3(exprMatr, nCores = 4, verbose = TRUE)
linkList <- getLinkList(weightMat)
dim(linkList)
## [1] 380 3
head(linkList)
## regulatoryGene targetGene weight
## 1 Gene16 Gene18 0.2289570
## 2 Gene1 Gene6 0.2188915
## 3 Gene7 Gene18 0.1903439
## 4 Gene2 Gene19 0.1902978
## 5 Gene20 Gene13 0.1741122
## 6 Gene6 Gene1 0.1707038
linkList <- getLinkList(weightMat, reportMax = 5)
linkList <- getLinkList(weightMat, threshold = 0.1)
��ϸ��ת¼�� pbmc
# install.packages('pbmc3k.SeuratData', repos='http://seurat.nygenome.org/',
# type = 'source')
library(pbmc3k.SeuratData)
library(Seurat)
data("pbmc")
pbmc <- pbmc3k.final
levels(Idents(pbmc))
## [1] "Naive CD4 T" "Memory CD4 T" "CD14+ Mono" "B" "CD8 T"
## [6] "FCGR3A+ Mono" "NK" "DC" "Platelet"
��������ѡ��һ��ϸ������Ϊ��Memory CD4 T������صĻ���,����ȡ�������:
��ϸ����������������:
set.seed(1314) # For reproducibility of results
weightMat <- GENIE3(exprMatr, nCores = 4) # with the default parameters
weightMat[1:5, 1:5]
## LDHB MALAT1 EEF1A1 CCR7 TPT1
## MALAT1 0.04333745 0.00000000 0.14770305 0.04201807 0.07073973
## EEF1A1 0.04337260 0.08230214 0.00000000 0.02626972 0.03591088
## CCR7 0.01151539 0.02685912 0.01375829 0.00000000 0.01048274
## TPT1 0.03177815 0.04102548 0.04016091 0.02505163 0.00000000
## CD3D 0.03271693 0.02794367 0.01997983 0.02297784 0.02406677
�������������ӻ�
������ֵĿ��ӻ���������
library(igraph)
weightMat[which(weightMat < 0.04)] = 0
net1 <- graph_from_incidence_matrix(weightMat)
layouts <- grep("^layout_", ls("package:igraph"), value = TRUE)[-1]
# Remove layouts that do not apply to our graph.
layouts <- layouts[!grepl("bipartite|merge|norm|sugiyama|tree", layouts)]
layouts <- c("layout_as_star", "layout_in_circle", "layout_nicely", "layout_on_grid",
"layout_on_sphere", "layout_randomly", "layout_with_dh", "layout_with_drl", "layout_with_fr",
"layout_with_gem", "layout_with_graphopt", "layout_with_kk", "layout_with_lgl",
"layout_with_mds")
layouts
## [1] "layout_as_star" "layout_in_circle" "layout_nicely"
## [4] "layout_on_grid" "layout_on_sphere" "layout_randomly"
## [7] "layout_with_dh" "layout_with_drl" "layout_with_fr"
## [10] "layout_with_gem" "layout_with_graphopt" "layout_with_kk"
## [13] "layout_with_lgl" "layout_with_mds"
length(layouts)
## [1] 14
par(mfrow = c(3, 5), mar = c(1, 1, 1, 1))
for (layout in layouts) {
print(layout)
l <- do.call(layout, list(net1))
plot(net1, edge.arrow.mode = 1, layout = l, main = layout)
}
## [1] "layout_as_star"
## [1] "layout_in_circle"
## [1] "layout_nicely"
## [1] "layout_on_grid"
## [1] "layout_on_sphere"
## [1] "layout_randomly"
## [1] "layout_with_dh"
## [1] "layout_with_drl"
## [1] "layout_with_fr"
## [1] "layout_with_gem"
## [1] "layout_with_graphopt"
## [1] "layout_with_kk"
## [1] "layout_with_lgl"
## [1] "layout_with_mds"

�����ǵĹ�����,�����������������û���κε�����,������û��ʲô���ع�ϵ,�����뿴��Щ�������е��ع�ϵ�ġ�
V(net1)[igraph::degree(net1) > 1]
## + 26/118 vertices, named, from ec4a27c:
## [1] MALAT1 EEF1A1 TPT1 NPM1 EEF1B2 JUNB TMEM66 BTG1 LDHB
## [10] LDHB MALAT1 EEF1A1 CCR7 TPT1 NPM1 NOSIP LEF1 EEF1B2
## [19] JUNB TMEM66 GLTSCR2 BTG1 IL7R MAL LCK SOCS3
plot(induced_subgraph(net1, V(net1)[igraph::degree(net1) > 1]))
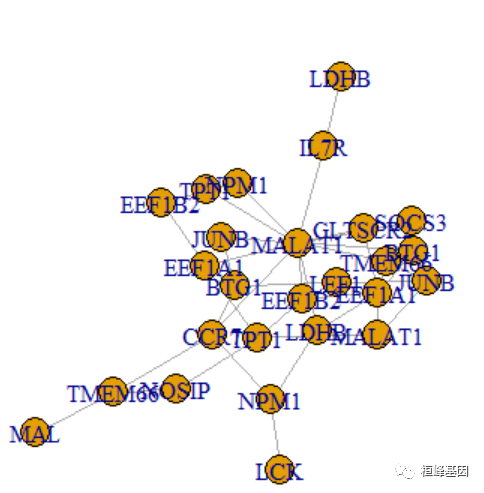
GENIE3 ���ǵ��ܶ�ͯЬ����������ܻ�������,�Ͱ�����һ���������ݸ�������ֵɾѡ
linkList <- getLinkList(weightMat, reportMax = 10)
linkList <- getLinkList(weightMat, threshold = 0.01)
head(linkList)
## regulatoryGene targetGene weight
## 1 MALAT1 EEF1A1 0.14770305
## 2 EEF1A1 MALAT1 0.08230214
## 3 BTG1 JUNB 0.07229212
## 4 MALAT1 TPT1 0.07073973
## 5 JUNB BTG1 0.06944210
## 6 MALAT1 EEF1B2 0.06485446
�����ʽ��ȻҲ�ǿ�����igraph����������ġ�
net <- graph_from_data_frame(linkList)
plot(net, edge.arrow.size = 0.2, edge.curved = 0, vertex.color = "orange", vertex.frame.color = "#555555",
vertex.label.color = "black", vertex.label.cex = 0.7)
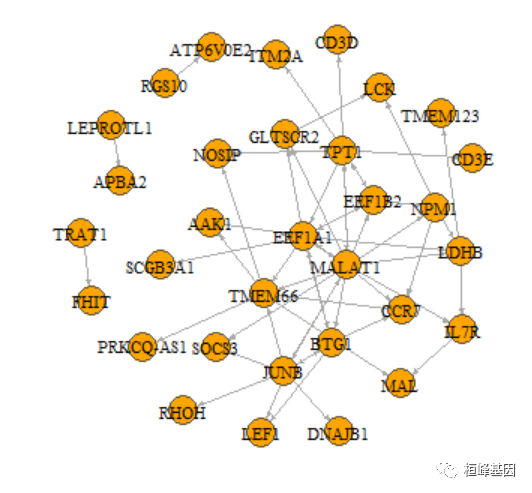
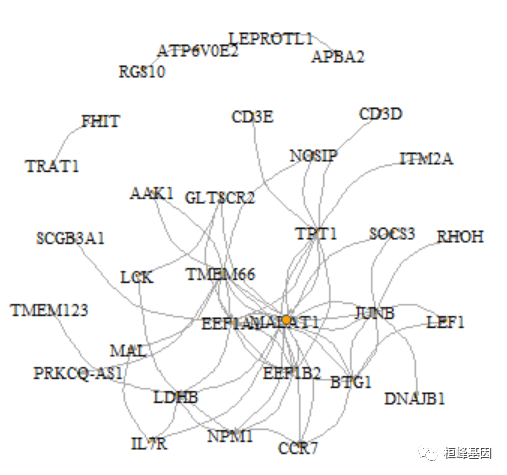
���ǰѱ�������Ϣ��������ͼ��:
avexp <- AverageExpression(cd4nt, features = names(V(net)), slot = "data")
V(net)$size <- abs(scale(avexp$RNA))
E(net)$width <- E(net)$weight * 10
plot(net, edge.arrow.size = 0.2, edge.curved = 0.5, vertex.color = "orange", vertex.frame.color = "#555555",
vertex.label.color = "black", vertex.label.cex = 0.7)
����������Ҫ���ܻ���ת¼�����ݵĻ�����������ƶ� (GENIE3)��Ŀǰ��ϸ������ķ���Ҳ�ڽ���,��ϸ��ϵ�п�����Ŀǰ�IJ�������,���ⷽ���������ʦ,��ϵ�������,�ṩ��߶˵Ŀ��з���!
�������,����ɹ�����!
δ����������ںŽ�����ϵ��Ƴ���ϸ��ϵ�����ŷ����̳�,
�����ڴ�!!
��������Ž���Ⱥ����ʦ����ɨ���һ����ά�����,��ע����λ+����+Ŀ�ġ�,��Щ�뷢���ľ�����Ű�,���÷����������߳�ȥ!
References:
-
Aibar, Sara, et al. 2017. ��SCENIC: Single-Cell Regulatory Network Inference and Clustering.�� Nature Methods 14 (october): 1083�C6. doi:10.1038/nmeth.4463.
-
Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P (2010) Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 5(9): e12776.