Co-occurrence网络图绘制教程(附详细代码)
前段时间阅读微生物生态学的文章总能看见有关co-occrrence这类的网络图,用来表示ARG的潜在宿主。例如下面这张图:
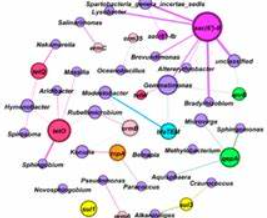
第一次看到这张图的时候感觉好厉害的,后来自己到处问问也打听到了相关的绘制方法。方法有很多种,但是本文用的就是R和Gephi。之前见《宏基因组》公众号中也出过相关的教程,但是好像很多人做到一半就卡壳了。之前也尝试去复现了一下,发现没有成功。后来自己整理了一下这个过程,主要分为两个部分:1、使用R语言获得网络图源数据;2、使用Gephi对得到的图形源数据进行可视化。
R语言计算网络图源数据
code读入data的样式(行表示不同的样本,列表示不同种类的属级微生物):

完整code:
data <- read.csv('data.csv',row.names=1)#读入数据
speciesandgene <- t(combn(colnames(data),2))#得到所有属级微生物的排列组合结果即为两列数据
library(Hmisc)#使用Hmisc包
randp <- rcorr(as.matrix(data),type = 'pearson')#使用rcorr函数计算相关系数,至于为什么使用这个包详见这篇博客https://blog.csdn.net/yangqijia1/article/details/119874420?spm=1001.2014.3001.5502
r <- randp$r#得到相关系数的矩阵
p <- randp$P
r[!lower.tri(r,diag = F)] <- NA#获取下三角矩阵并把对角线的值去掉
r <- matrix(c(r))
r <- matrix(r[complete.cases(r),])#把相关系数矩阵转化为一列数据
p[!lower.tri(p,diag = F)] <- NA#P值的处理过程和R一致,最后得到一列数据
p <- matrix(c(p))
p <- matrix(p[complete.cases(p),])
res <- cbind(speciesandgene,r,p)#把第二行的结果(所有的微生物排列组合结果)和相关系数值、P值组合在一起
res <- subset(res,res[,3]>=0.6&res[,4]<=0.05)#按照一定的阈值对结果进行筛选
write.csv(res,file = 'res.csv',row.names = F)#把结果导出为csv格式的表格
以上code得到的未筛选结果(第一二列对应不同微生物的排列组合结果,第三列为相关系数值,第四列为P值):

最后就是利用得到这个数据进行可视化即可
Gephi软件进行可视化
Gephi软件的可视化教程详见B站:https://www.bilibili.com/video/BV1cB4y1N77j/,各位有问题可以B站后台私信up主(人敲好的!)。